应用案例
本章将介绍AuToFF结合其他的分子动力学模拟软件的相关MD应用案例,并针对具体的算例给出基本的使用说明。
锂离子电池离子液体电解质体系的MD模拟
锂电池因其较高能量密度受到学术界和工业界的广泛关注。电解液作为锂电池的一个关键组成,对锂电池性能具有十分重要的影响。离子液体是由有机阳离子以及有机或无机阴离子所组成的室温有机熔融盐,具有低的挥发性、不可燃、高的热以及化学稳定性、宽的电化学窗口、高的离子电导率以及结构可调谐等优势,得到了广泛的研究与应用。
力场辅助工具-AuToFF
确定起始构型
确定双(氟磺酰)亚胺(FSI)阴离子结构,可选择点击AuToFF程序-离子液体模块进行2D建模(如图1),点击“ 生成3d结构视图 ”按钮即可3D显示。

图3.1.1 创建FSI分子结构
完成分子建模后,可以支持多种结构文件类型下载,包括.pdb、.mol、.mol2、.xyz
图3.1.2 下载结构文件
下载FSI分子的pdb结构文件如下:
TITLE bis(fluorosulfonyl)imide
ATOM 1 NBT NS 1 -0.168 -0.820 -0.081 1.00 0.00
ATOM 2 SBT NS 1 -1.632 -0.300 -0.128 1.00 0.00
ATOM 3 SBT NS 1 0.960 0.020 -0.721 1.00 0.00
ATOM 4 OBT NS 1 0.879 0.465 -2.120 1.00 0.00
ATOM 5 OBT NS 1 1.575 0.970 0.179 1.00 0.00
ATOM 6 OBT NS 1 -2.303 -0.173 -1.425 1.00 0.00
ATOM 7 OBT NS 1 -2.428 -1.028 0.848 1.00 0.00
ATOM 8 F1 NS 1 2.045 -1.173 -0.898 1.00 0.00
ATOM 9 F1 NS 1 -1.649 1.230 0.367 1.00 0.00
END
同理,建立了THF分子结构如下:

图3.1.3 创建THF分子结构
THF分子的pdb结构文件如下:
ATOM 1 O1 THF 1 1.138 -0.023 -0.012 1.00 0.00 O
ATOM 2 C2 THF 1 0.289 -1.168 0.135 1.00 0.00 C
ATOM 3 C3 THF 1 -1.119 -0.718 -0.192 1.00 0.00 C
ATOM 4 C4 THF 1 -1.096 0.731 0.223 1.00 0.00 C
ATOM 5 C5 THF 1 0.316 1.147 -0.124 1.00 0.00 C
ATOM 6 H6 THF 1 0.365 -1.509 1.173 1.00 0.00 H
ATOM 7 H7 THF 1 0.635 -1.970 -0.522 1.00 0.00 H
ATOM 8 H8 THF 1 -1.883 -1.309 0.317 1.00 0.00 H
ATOM 9 H9 THF 1 -1.292 -0.791 -1.273 1.00 0.00 H
ATOM 10 H10 THF 1 -1.254 0.808 1.305 1.00 0.00 H
ATOM 11 H11 THF 1 -1.854 1.341 -0.275 1.00 0.00 H
ATOM 12 H12 THF 1 0.696 1.928 0.541 1.00 0.00 H
ATOM 13 H13 THF 1 0.383 1.506 -1.157 1.00 0.00 H
END
同理,建立了TTE分子结构如下:
图3.1.4 创建TTE分子结构
TTE分子的pdb结构文件如下:
ATOM 1 C1 TTE 1 1.142 0.720 -2.222 1.00 0.00 C
ATOM 2 C2 TTE 1 0.567 -0.382 -1.346 1.00 0.00 C
ATOM 3 C3 TTE 1 1.027 -1.774 -1.781 1.00 0.00 C
ATOM 4 O4 TTE 1 2.454 -1.899 -1.714 1.00 0.00 O
ATOM 5 C5 TTE 1 2.882 -3.210 -2.100 1.00 0.00 C
ATOM 6 C6 TTE 1 4.403 -3.383 -2.012 1.00 0.00 C
ATOM 7 F7 TTE 1 4.779 -4.634 -2.394 1.00 0.00 F
ATOM 8 F8 TTE 1 0.935 -0.190 -0.047 1.00 0.00 F
ATOM 9 F9 TTE 1 -0.795 -0.353 -1.362 1.00 0.00 F
ATOM 10 F10 TTE 1 0.773 0.545 -3.518 1.00 0.00 F
ATOM 11 F11 TTE 1 0.660 1.937 -1.845 1.00 0.00 F
ATOM 12 F12 TTE 1 2.261 -4.100 -1.283 1.00 0.00 F
ATOM 13 F13 TTE 1 2.429 -3.437 -3.358 1.00 0.00 F
ATOM 14 F14 TTE 1 5.042 -2.519 -2.846 1.00 0.00 F
ATOM 15 H15 TTE 1 2.232 0.764 -2.170 1.00 0.00 H
ATOM 16 H16 TTE 1 0.683 -1.969 -2.803 1.00 0.00 H
ATOM 17 H17 TTE 1 0.564 -2.514 -1.120 1.00 0.00 H
ATOM 18 H18 TTE 1 4.767 -3.208 -0.996 1.00 0.00 H
END
还建立了 \(\ce{Li^+}\) 结构, \(\ce{Li^+}\) 的pdb结构文件如下:
REMARK 1 File created by GaussView 6.0.16
HETATM 1 Li 0 -1.265 -0.267 0.000 Li
END
选用适当力场和模拟软件
选择适当的力场是进行MD模拟的基础,可以快速地获得准确的模拟结果。针对离子液体FSI选择OPLS力场即可,确定原子类型
图3.1.5 根据力场选择原子类型
Note
点击结构视图中原子可进行配置原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件的相关力场参数,包括LJ、键、键角、二面角参数,原子电荷。此外生成拓扑文件可支持多款计算软件,包括:GROMACS、LAMMPS、AMBER、Moltemplate、OpenMM、TINKER、CHARMM。下载的文件夹中除了力场拓扑文件之外还包含力场参数的文献来源。
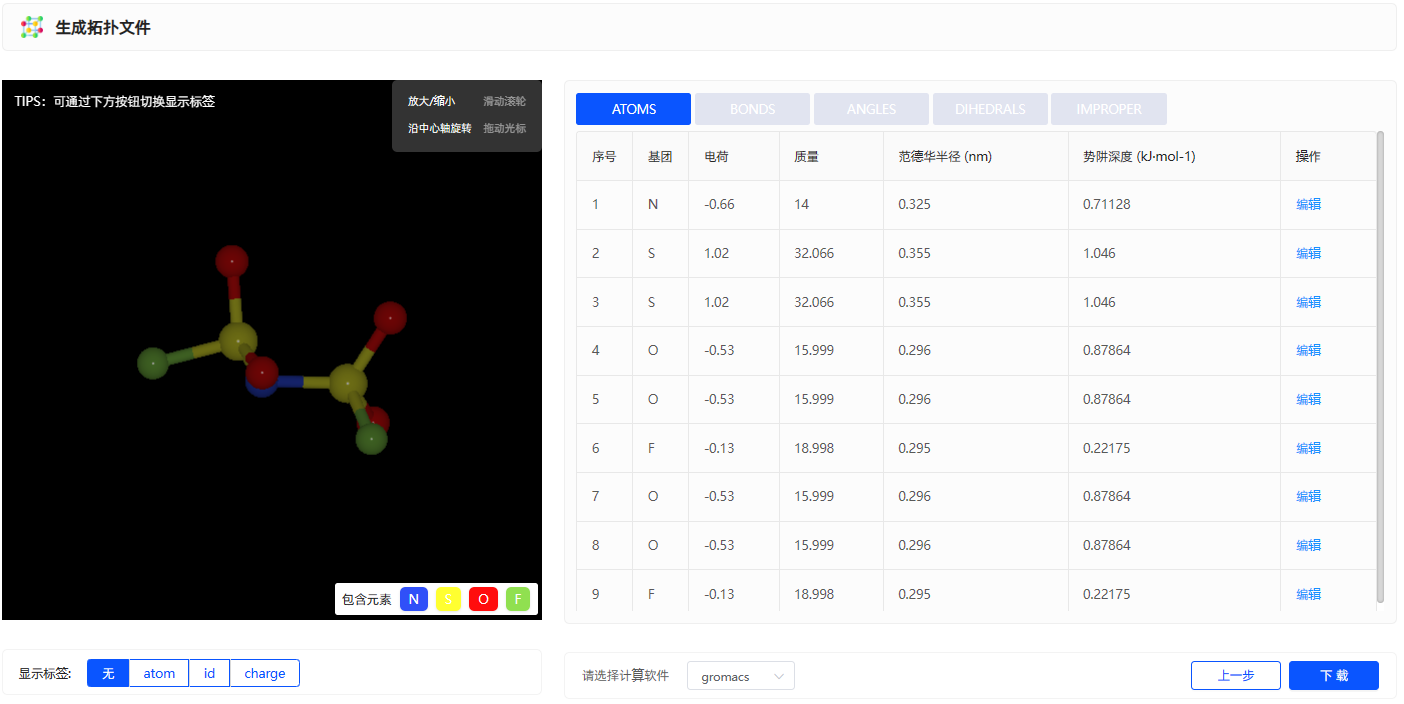
图3.1.6 生成拓扑文件
Note
点击下方显示标签按钮即可显示元素名称、原子ID、原子电荷。
用户也可通过 编辑 按钮进行自行修改力场参数信息。
模拟体系建模
构建体系
首先,创建模拟体系。通过Packmol软件,我们将离子液体的组成分子放入一个立方体的模拟盒子中。这个过程中立方体的盒子大小要略大于同等密度下离子液体所需要的体积,以保证有足够的空间使得离子液体分子能够随机的分布并且模拟可以快速平衡。将AuToFF创建并下载好每个组分的拓扑文件,然后把pdb文件拷贝到packmol文件夹,调用packmol程序生成模拟的盒子。Packmol输入文件model.inp如下:
tolerance 2.0
filetype pdb
add_box_sides 1.5
output model.pdb
structure Li.pdb
number 63
inside cube 0. 0. 0. 60
end structure
structure FSI.pdb
number 63
inside cube 0. 0. 0. 60
end structure
structure THF.pdb
number 310
inside cube 0. 0. 0. 60
end structure
structure TTE.pdb
number 165
inside cube 0. 0. 0. 60
end structure
运行 packmol < model.inp 可生成model.pdb文件,该文件包含了锂离子离子液体电解质模拟体系中所有原子的坐标,但缺少键、键角等拓扑结构信息。将得到的model.pdb导入到VMD显示如下

图3.1.7 模拟体系初始构型
构建拓扑文件
拓扑文件是gromacs运行模拟所必需的文件,它提供了模拟体系中所有分子的拓扑结构、力场文件的引用、约束力参数……;拓扑文件必须包含三个层次:
参数级别;这一部分包括了力场设定
分子定义级别:这一部分包含了一个或多个分子对应的.itp文件。实际上,.itp 文件可以看做是 .top 文件分子定义级别(针对每单个分子)单独拿出储存的信息,他们形成了一个嵌套式的引用关系
体系级别:只包含体系的特定信息
锂离子离子液体电解质模拟体系的top文件model.top如下:
#define _FF_OPLS
#define _FF_OPLSAA
[ defaults ]
1 3 yes 0.5 0.5
#include "Li_ATP.itp"
#include "FSI_ATP.itp"
#include "THF_ATP.itp"
#include "TTE_ATP.itp"
#include "Li.itp"
#include "FSI.itp"
#include "THF.itp"
#include "TTE.itp"
[ system ]
63Li+63FSI+310THF+165TTE
[ molecules ]
Li 63
FSI 63
THF 310
TTE 165
MD模拟
能量最小化
随后通过共轭梯度法优化初始结构,使得分子间的距离合适,没有较大的应力。gromacs能量最小化em.mdp输入如下:
define = -DFLEXIBLE
integrator = cg
nsteps = 10000
emtol = 100.0
emstep = 0.01
;
nstxout = 100
nstlog = 50
nstenergy = 50
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
DispCorr = EnerPres
;
constraints = none
MD平衡过程
在模拟过程中,模拟步长设为2fs,采用Verlet算法来计算运动方程。模拟体系的三个方向均考虑周期性,是体相的模拟。为了使模拟体系快速合理达到平衡状态,采用梯度退火模拟。具体流程如下:等温等压系综下,模拟体系首先被缓慢加热到500 K,并在500 K下维持1 ns的NPT系综模拟,然后逐步将温度下降至400K ,并在400 K下维持1 ns的NPT系综模拟,最后再逐步将温度下降至目标温度298.15 K。当体系温度达到模拟的目标温度后,继续保持NPT系综计算2 ns,以保证模拟体系的能量、密度的性质趋于收敛,体系保持平衡。gromacs平衡过程eq.mdp输入如下:
define =
integrator = md
dt = 0.002
nsteps = 5000000
comm-grps = system
energygrps =
;
nstxout = 0
nstvout = 0
nstfout = 0
nstlog = 500
nstenergy = 500
nstxout-compressed = 1000
compressed-x-grps = system
;
annealing = single
annealing_npoints = 7
annealing_time = 0 1000 2000 3000 4000 5000 7000
annealing_temp = 0 500 500 400 400 298.15 298.15
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = cut-off
rvdw = 1.0
DispCorr = EnerPres
;
Tcoupl = V-rescale
tau_t = 0.5
tc_grps = system
ref_t = 298.15
;
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 1
ref_p = 1.01325
compressibility = 8.5e-5
;
gen_vel = no
gen_temp = 298.15
gen_seed = -1
;
freezegrps =
freezedim =
constraints = hbonds
MD采样过程
最后,在体系平衡的基础上,继续模拟2 ns ,并采样、分析、计算体系结构和性质等信息。gromacs模拟计算prod.mdp输入如下:
define =
integrator = md
dt = 0.002
nsteps = 1000000
comm-grps = system
energygrps =
;
nstxout = 0
nstvout = 0
nstfout = 0
nstlog = 500
nstenergy = 500
nstxout-compressed = 1000
compressed-x-grps = system
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = cut-off
rvdw = 1.0
DispCorr = EnerPres
;
Tcoupl = V-rescale
tau_t = 0.5
tc_grps = system
ref_t = 298.15
;
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 1
ref_p = 1.01325
compressibility = 8.5e-5
;
gen_vel = no
gen_temp = 298.15
gen_seed = -1
;
freezegrps =
freezedim =
constraints = hbonds
MD结果分析
模拟平衡结构快照图
取出模拟平衡后最后一帧结构,导入VMD即可查看快照图如下:
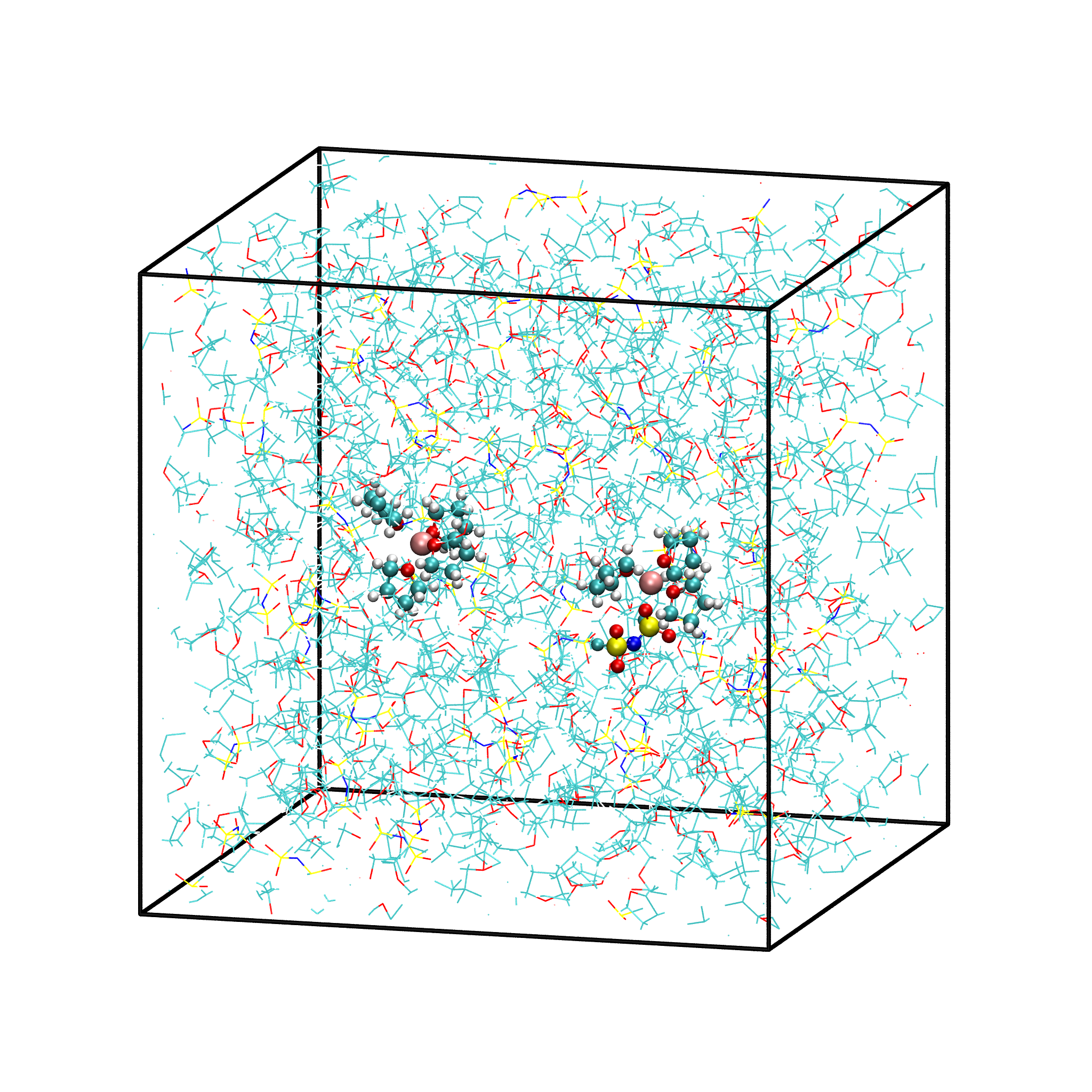
图3.1.8 模拟平衡结构快照图
Note
gromacs转换成pdb结构文件命令: gmx trjconv -f prod.xtc -s prod.tpr -o prod.pdb -dump 2000
径向分布函数(RDF)
为了研究体系的局部结构特征,统计体系径向分布函数,计算 \(\ce{Li^+}\) 的配位数,
其中,\(\ce{r_{min}}\) 为径向分布函数中第一波谷对应的位置, \({\rho_𝛽}\) 为体系中平均粒子密度。

图3.1.9 径向分布函数图
Note
gromacs可以生成径向分布函数,命令为:gmx rdf -f prod.xtc -s prod.tpr -o rdf.xvg -cn rdf_cn.xvg -bin 0.005 -b 1000 -e 2000 -rmax 1
均方位移(MSD)和扩散系数
为了探究 \(\ce{Li^+}\) 的扩散系数,gromacs可计算均方位移,模拟了不同温度下离子的扩散性质,如下图:
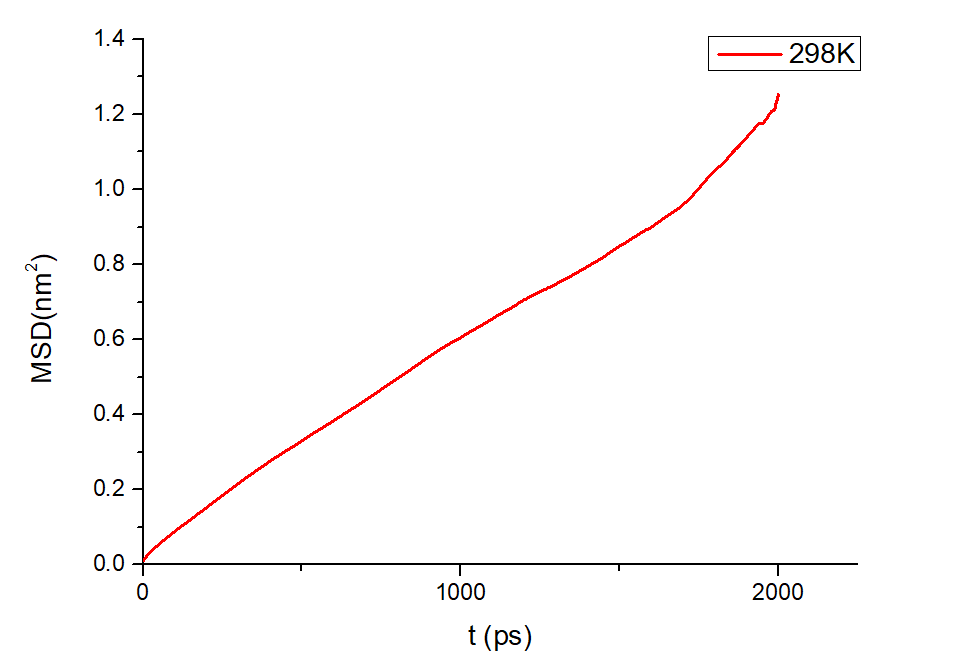
图3.1.10 均方位移图
Note
gromacs可以计算均方位移,命令为:gmx msd -f eq.xtc -s eq.tpr -beginfit 830 -endfit 1400 -trestart 0.002
继而可通过平衡分子动力学(EMD)模拟计算扩散系数,粒子的自扩散系数与其均方位移对时间的导数有关
计算所得,298K温度下 \(\ce{Li^+}\) 的扩散系数为0.0812 (+/- 0.0113) 1e-5 \(\ce{(cm^2/s)}\)
锂离子电池聚合物电解质体系的MD模拟
锂离子电池因能量密度高、循环稳定性好、无污染等优点已广泛应用于小型电子器件和电动汽车上,并且有望成为未来大型储能系统(EES)的储电供电设备。其中,聚合物电解质具有易加工性、改善锂枝晶生长和提高电池安全性等优点。
力场辅助工具-AuToFF
确定起始构型
确定双(氟磺酰)亚胺(FSI)阴离子结构,可选择点击AuToFF程序-离子液体模块进行2D建模(如图1),点击“ 生成3d结构视图 ”按钮即可3D显示。
图3.2.1 创建FSI结构
完成分子建模后,可以支持多种结构文件类型下载,包括.pdb、.mol、.mol2、.xyz
图3.2.2 下载结构文件
下载FSI分子的pdb结构文件如下:
TITLE bis(fluorosulfonyl)imide
ATOM 1 NBT NS 1 -0.168 -0.820 -0.081 1.00 0.00
ATOM 2 SBT NS 1 -1.632 -0.300 -0.128 1.00 0.00
ATOM 3 SBT NS 1 0.960 0.020 -0.721 1.00 0.00
ATOM 4 OBT NS 1 0.879 0.465 -2.120 1.00 0.00
ATOM 5 OBT NS 1 1.575 0.970 0.179 1.00 0.00
ATOM 6 OBT NS 1 -2.303 -0.173 -1.425 1.00 0.00
ATOM 7 OBT NS 1 -2.428 -1.028 0.848 1.00 0.00
ATOM 8 F1 NS 1 2.045 -1.173 -0.898 1.00 0.00
ATOM 9 F1 NS 1 -1.649 1.230 0.367 1.00 0.00
END
还建立了 \(\ce{Li^+}\) 结构, \(\ce{Li^+}\) 的pdb结构文件如下:
REMARK 1 File created by GaussView 6.0.16
HETATM 1 Li 0 -1.265 -0.267 0.000 Li
END
此外,AuToFF可直接进行聚合物建模,建立PEO聚合物。如下:

图3.2.3 创建PEO结构
聚合物PEO结构文件下载链接 PEO.pdb
选用适当力场和模拟软件
选择适当的力场是进行MD模拟的基础,可以快速地获得准确的模拟结果。针对离子液体FSI选择OPLS力场即可,确定原子类型
图3.2.4 根据力场选择原子类型
Note
点击结构视图中原子可进行配置原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件的相关力场参数,包括LJ、键、键角、二面角参数,原子电荷。此外生成拓扑文件可支持多款计算软件,包括:GROMACS、LAMMPS、AMBER、Moltemplate、OpenMM、TINKER、CHARMM、RASPA、NAMD、GOMC。下载的文件夹中除了力场拓扑文件之外还包含力场参数的文献来源。
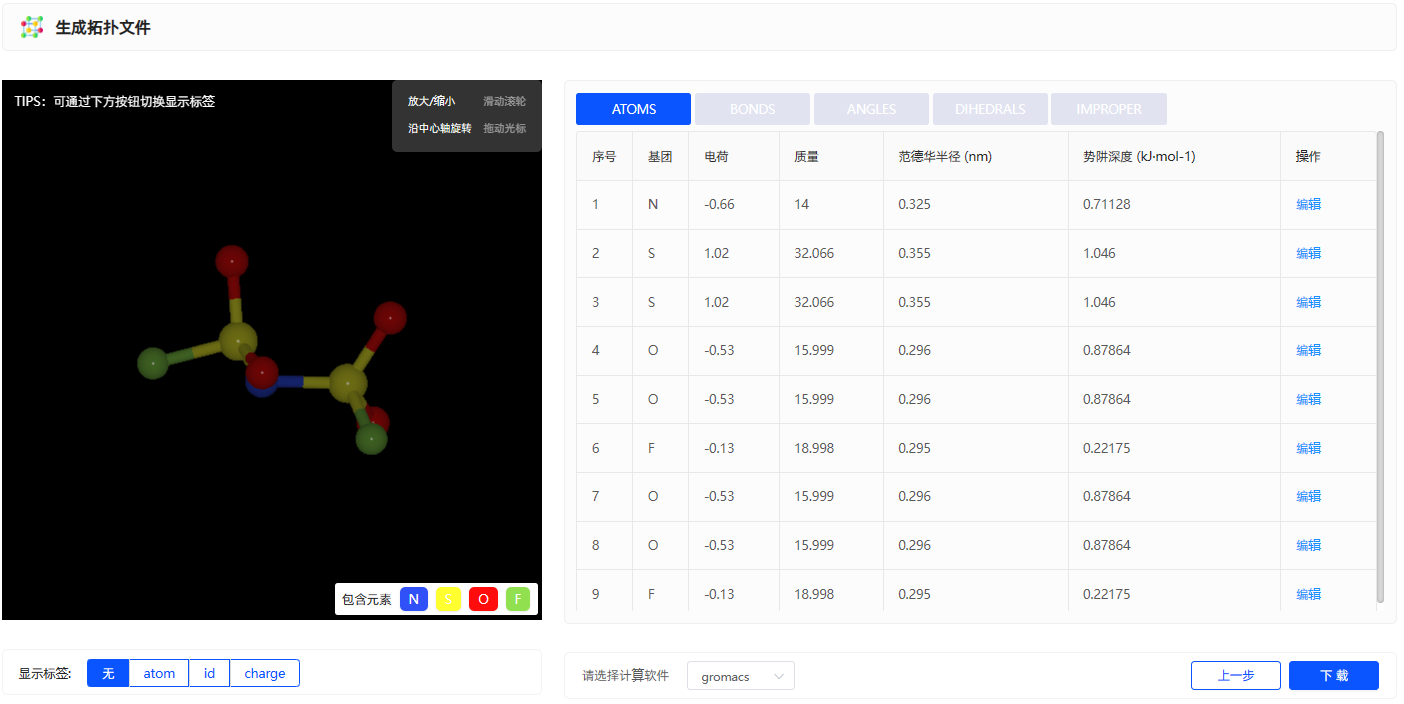
图3.2.5 生成拓扑文件
Note
点击下方显示标签按钮即可显示元素名称、原子ID、原子电荷。
用户也可通过 编辑 按钮进行自行修改力场参数信息。
模拟体系建模
构建体系
首先,创建模拟体系。通过Packmol软件,我们将聚合物电解质体系的组成分子放入一个立方体的模拟盒子中。这个过程中立方体的盒子大小要略大于同等密度下聚合物电解质体系所需要的体积,以保证有足够的空间使得模拟体系内分子能够随机的分布并且模拟可以快速平衡。Packmol输入文件model.inp如下:
tolerance 2.0
filetype pdb
add_box_sides 1.5
output model.pdb
structure Li.pdb
number 500
inside cube 0. 0. 0. 150
end structure
structure FSI.pdb
number 500
inside cube 0. 0. 0. 150
end structure
structure PEO.pdb
number 40
inside cube 0. 0. 0. 150
end structure
运行 packmol < model.inp 可生成model.pdb文件,该文件包含了锂离子聚合物电解质模拟体系中所有原子的坐标,但缺少键、键角等拓扑结构信息。将得到的model.pdb导入到VMD显示如下

图3.2.6 模拟体系初始构型
构建拓扑文件
拓扑文件是gromacs运行模拟所必需的文件,它提供了模拟体系中所有分子的拓扑结构、力场文件的引用、约束力参数……;拓扑文件必须包含三个层次:
参数级别;这一部分包括了力场设定
分子定义级别:这一部分包含了一个或多个分子对应的.itp文件。实际上,.itp 文件可以看做是 .top 文件分子定义级别(针对每单个分子)单独拿出储存的信息,他们形成了一个嵌套式的引用关系
体系级别:只包含体系的特定信息
锂离子聚合物电解质模拟体系的top文件model.top如下:
#define _FF_OPLS
#define _FF_OPLSAA
[ defaults ]
1 3 yes 0.5 0.5
#include "PEO_ATP.itp"
#include "Li_ATP.itp"
#include "FSI_ATP.itp"
#include "PEO.itp"
#include "Li.itp"
#include "FSI.itp"
[ system ]
25DSPE+5AIE
[ molecules ]
PEO 40
Li 500
FSI 500
MD模拟
能量最小化
随后通过共轭梯度法优化初始结构,使得分子间的距离合适,没有较大的应力。gromacs能量最小化em.mdp输入如下:
define = -DFLEXIBLE
integrator = cg
nsteps = 10000
emtol = 100.0
emstep = 0.01
;
nstxout = 100
nstlog = 50
nstenergy = 50
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
DispCorr = EnerPres
;
constraints = none
MD平衡过程
在模拟过程中,模拟步长设为2fs,采用Verlet算法来计算运动方程。模拟体系的三个方向均考虑周期性,是体相的模拟。为了使模拟体系快速合理达到平衡状态,采用梯度退火模拟。具体流程如下:等温等压系综下,模拟体系首先被缓慢加热到500 K,并在500 K下维持1 ns的NPT系综模拟,然后逐步将温度下降至400K ,并在400 K下维持1 ns的NPT系综模拟,最后再逐步将温度下降至目标温度298.15 K。当体系温度达到模拟的目标温度后,继续保持NPT系综计算2 ns,以保证模拟体系的能量、密度的性质趋于收敛,体系保持平衡。gromacs平衡过程eq.mdp输入如下:
define =
integrator = md
dt = 0.002
nsteps = 5000000
comm-grps = system
energygrps =
;
nstxout = 0
nstvout = 0
nstfout = 0
nstlog = 500
nstenergy = 500
nstxout-compressed = 1000
compressed-x-grps = system
;
annealing = single
annealing_npoints = 7
annealing_time = 0 1000 2000 3000 4000 5000 7000
annealing_temp = 0 500 500 400 400 298.15 298.15
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = cut-off
rvdw = 1.0
DispCorr = EnerPres
;
Tcoupl = V-rescale
tau_t = 0.5
tc_grps = system
ref_t = 298.15
;
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 1
ref_p = 1.01325
compressibility = 8.5e-5
;
gen_vel = no
gen_temp = 298.15
gen_seed = -1
;
freezegrps =
freezedim =
constraints = hbonds
最后,在体系平衡的基础上,继续模拟2 ns ,并采样、分析、计算体系结构和性质等信息。gromacs模拟计算prod.mdp输入如下:
define =
integrator = md
dt = 0.002
nsteps = 1000000
comm-grps = system
energygrps =
;
nstxout = 0
nstvout = 0
nstfout = 0
nstlog = 500
nstenergy = 500
nstxout-compressed = 1000
compressed-x-grps = system
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = cut-off
rvdw = 1.0
DispCorr = EnerPres
;
Tcoupl = V-rescale
tau_t = 0.5
tc_grps = system
ref_t = 298.15
;
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 1
ref_p = 1.01325
compressibility = 8.5e-5
;
gen_vel = no
gen_temp = 298.15
gen_seed = -1
;
freezegrps =
freezedim =
constraints = hbonds
MD结果分析
模拟平衡结构快照图
取出模拟平衡后最后一帧结构,导入VMD即可查看快照图如下:
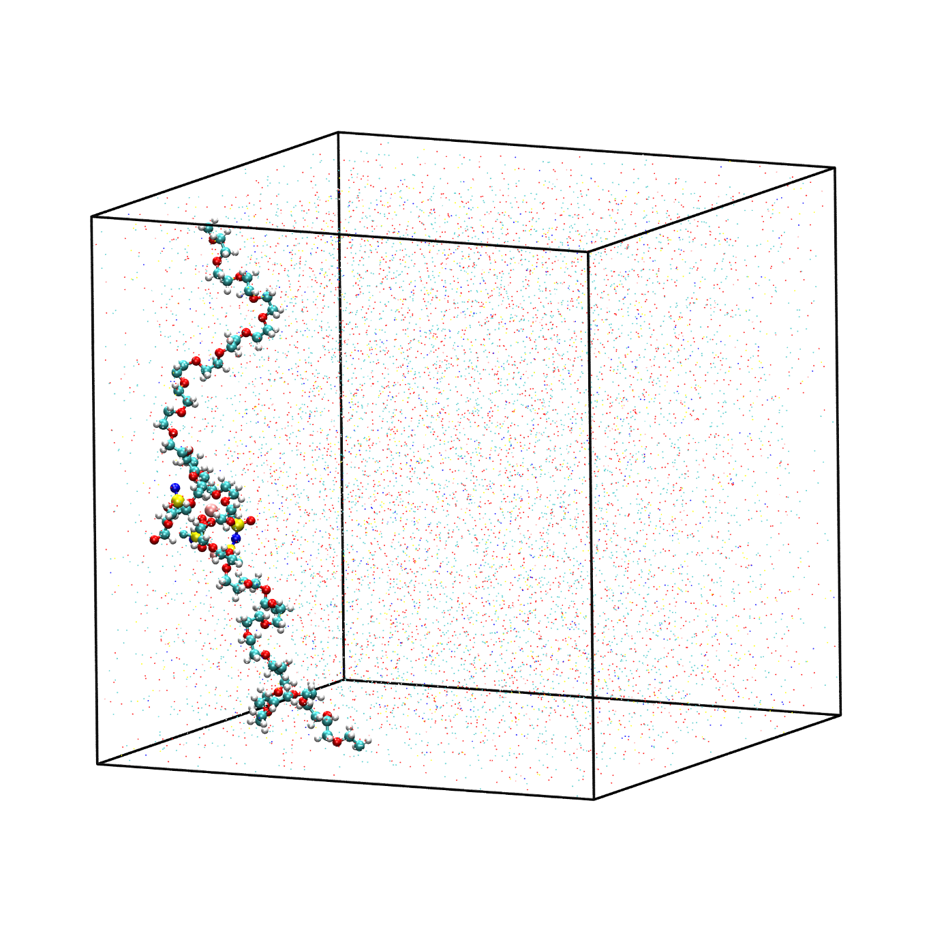
图3.2.7 模拟平衡结构快照图
Note
gromacs转换成pdb结构文件命令: gmx trjconv -f prod.xtc -s prod.tpr -o prod.pdb -dump 2000
径向分布函数(RDF)
为了研究体系的局部结构特征,统计体系径向分布函数,计算 \(\ce{Li^+}\) 的配位数,
其中,\(\ce{r_{min}}\) 为径向分布函数中第一波谷对应的位置, \({\rho_𝛽}\) 为体系中平均粒子密度。

图3.2.8 径向分布函数图
Note
gromacs可以生成径向分布函数,命令为:gmx rdf -f prod.xtc -s prod.tpr -o rdf.xvg -cn rdf_cn.xvg -bin 0.005 -b 1000 -e 2000 -rmax 1
均方位移(MSD)和扩散系数
为了探究 \(\ce{Li^+}\) 的扩散系数,gromacs可计算均方位移,模拟了不同温度下离子的扩散性质,如下图:
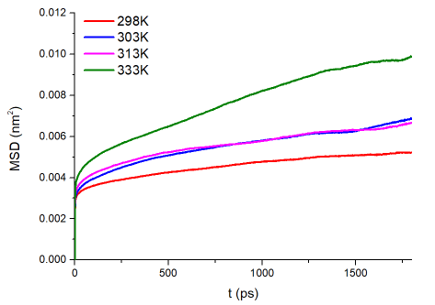
图3.2.9 均方位移图
Note
gromacs可以计算均方位移,命令为:gmx msd -f eq.xtc -s eq.tpr -beginfit 830 -endfit 1400 -trestart 0.002
继而可通过平衡分子动力学(EMD)模拟计算扩散系数,粒子的自扩散系数与其均方位移对时间的导数有关
结果如下表:
Temperature(K) |
\(\ce{𝐷_( Li^+ ) (cm^2/s)}\) |
298 |
0.0001587 (+/- 1.382e-05) 1e-5 |
303 |
0.0001887 (+/- 3.181e-05) 1e-5 |
313 |
0.0002174 (+/- 1.996e-05) 1e-5 |
333 |
0.0004883 (+/- 6.288e-05) 1e-5 |
冰块融化的MD模拟及TIP4P/ICE水模型使用
目前存在很多种水分子模型用于预测液态水的物理特性,或确定液态水的(未知)结构。AuToFF支持多种水模型,包括OPC, OPC3, SPC, SPC/E, SPC/Eb, TIP3P, TIPS3P, TIP3P-FB, TIP4P, TIP4P/Ew, TIP4P/2005, TIP4P/ICE, TIP4P/ε。其中,TIP4P/ICE是一种刚性平面的四点水模型,亦即除了一个O和2个H原子外还加入了一个虚原子M(在氧附近),虚原子仅具有负电荷,改善了水分子周围的静电分布。TIP4P/ICE是专门用来描述冰的一系列性质的水模型,对水的相图有较好的还原(1bar下冰-Ih熔点为272.2K),因此本案例将介绍利用TIP4P/ICE水模型进行冰融化的分子动力学模拟。
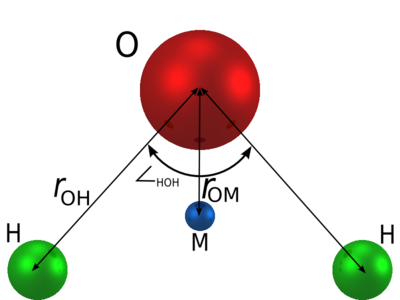
图3.3.1 TIP4P构型
接下来将介绍使用TIP4P/ICE水模型进行冰块融化的分子动力学模拟细节。
力场辅助工具-AuToFF
确定起始构型
选择AuToFF-全原子力场模块,上传冰晶体cif结构文件,水模型力场选择TIP4P/ICE,即可自动转成四点水模型的结构。
冰晶体的单胞结构文件下载链接 H2O-Ice-IV.cif
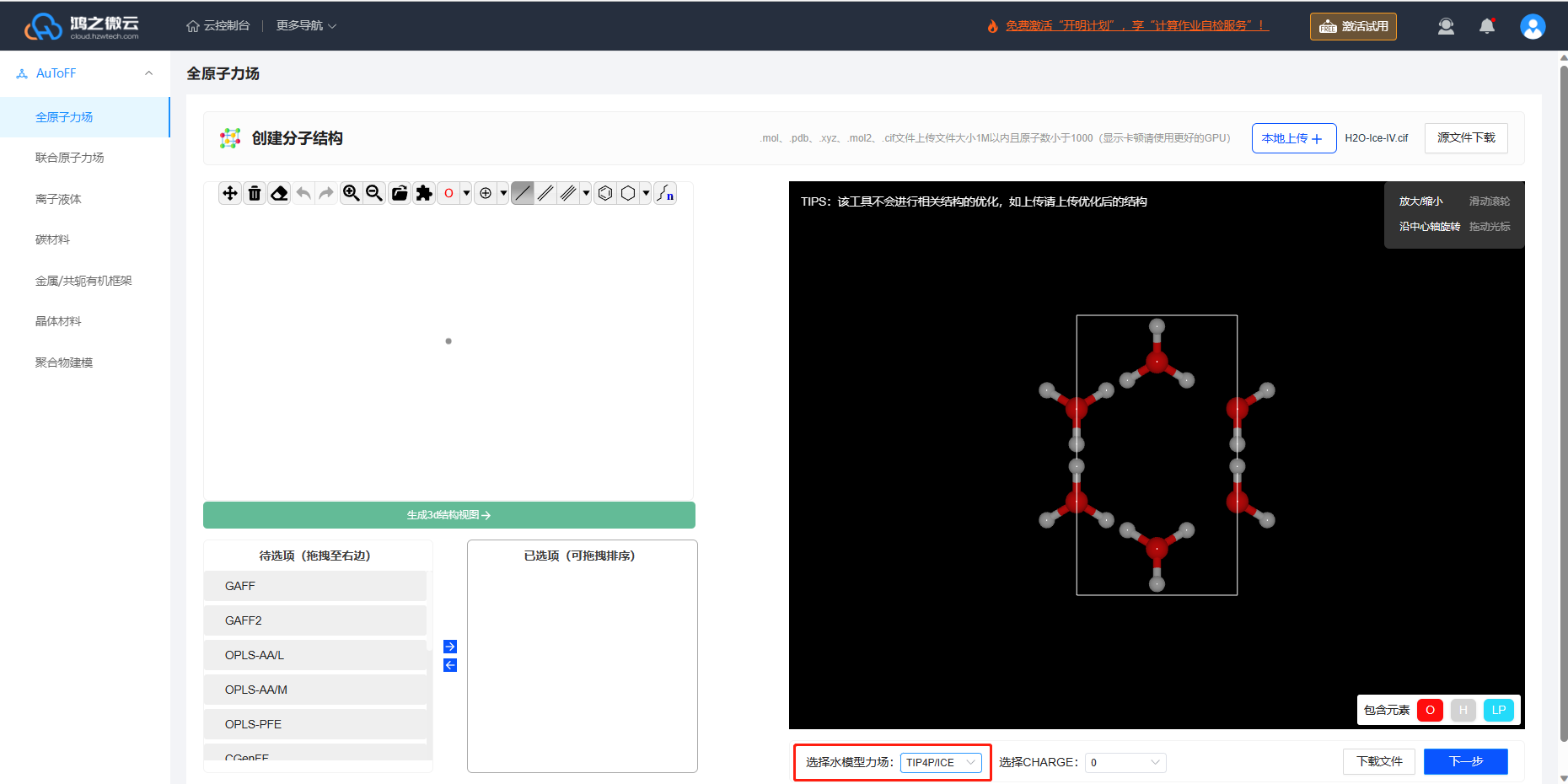
图3.3.2 确定冰晶体结构
选择力场
TIP4P/ICE是专门用来描述冰的一系列性质的水模型,对水的相图有较好的还原(1bar下冰-Ih熔点为272.2K),因此模拟冰融化过程也采用TIP4P/ICE力场模型。
图3.3.3 根据力场选择原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件(top文件)的相关力场参数(itp文件,包括LJ、键、键角参数,原子电荷)。此外生成拓扑文件可支持多款计算软件,包括:GROMACS、LAMMPS、AMBER、Moltemplate、OpenMM、TINKER、CHARMM。本案例推荐下载GROMACS的力场拓扑文件进行MD模拟,下载的文件夹中除了力场拓扑文件之外还包含力场参数的文献来源。针对晶体结构支持扩胞操作,此处设置的是5*5*5。
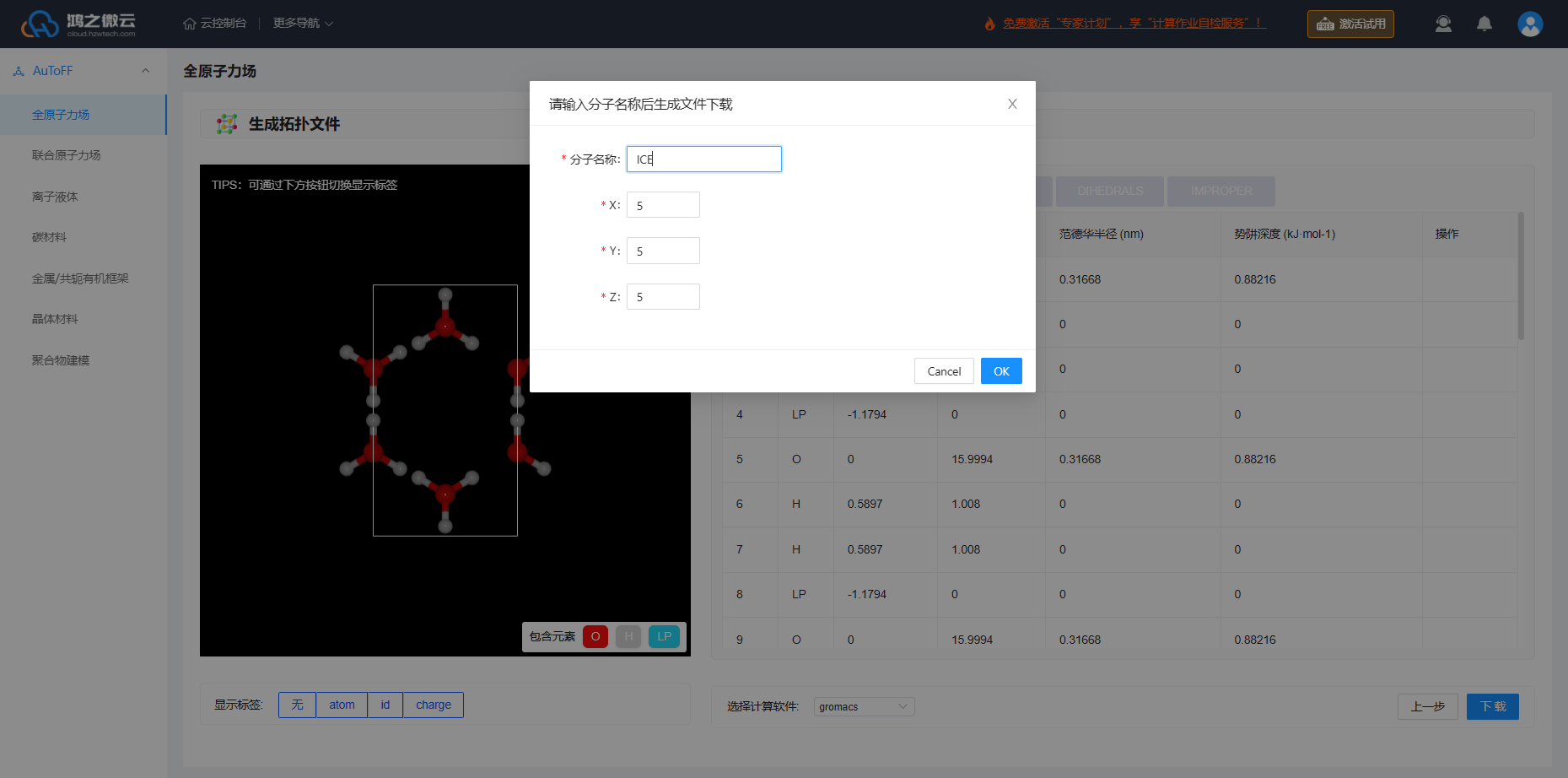
图3.3.4 生成冰晶体拓扑结构
冰晶体的结构文件下载链接 ICE.pdb
冰晶体模拟体系的top文件下载链接 ICE.top
MD模拟
在模拟过程中,模拟步长设为2fs,采用Verlet算法来计算运动方程。模拟体系的三个方向均考虑周期性,是体相的模拟。具体的冰融化过程模拟采用NPT系综,从0 K升温到350 K,使用V-rescale控温,参考温度298.15 K, Berendsen控压, 参考压力为 1.01325 bar 。完整的GROMACS的mdp文件输入如下:
define =
integrator = md
dt = 0.002
nsteps = 1000000
comm-grps = system
energygrps =
;
nstxout = 0
nstvout = 0
nstfout = 0
nstlog = 500
nstenergy = 500
nstxout-compressed = 1000
compressed-x-grps = system
;
annealing = single
annealing_npoints = 2
annealing_time = 0 2000 ;ps
annealing_temp = 0 350
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 0.9
vdwtype = cut-off
rvdw = 0.9
DispCorr = EnerPres
;
Tcoupl = V-rescale
tau_t = 0.2
tc_grps = system
ref_t = 298.15
;
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 0.5
ref_p = 1.01325
compressibility = 4.5e-5
;
gen_vel = no
gen_temp = 298.15
gen_seed = -1
;
freezegrps =
freezedim =
constraints = hbonds
MD结果分析
冰融化过程的轨迹变化通过VMD作图如下,可以清晰的展现出冰融化过程的体系结构变化。
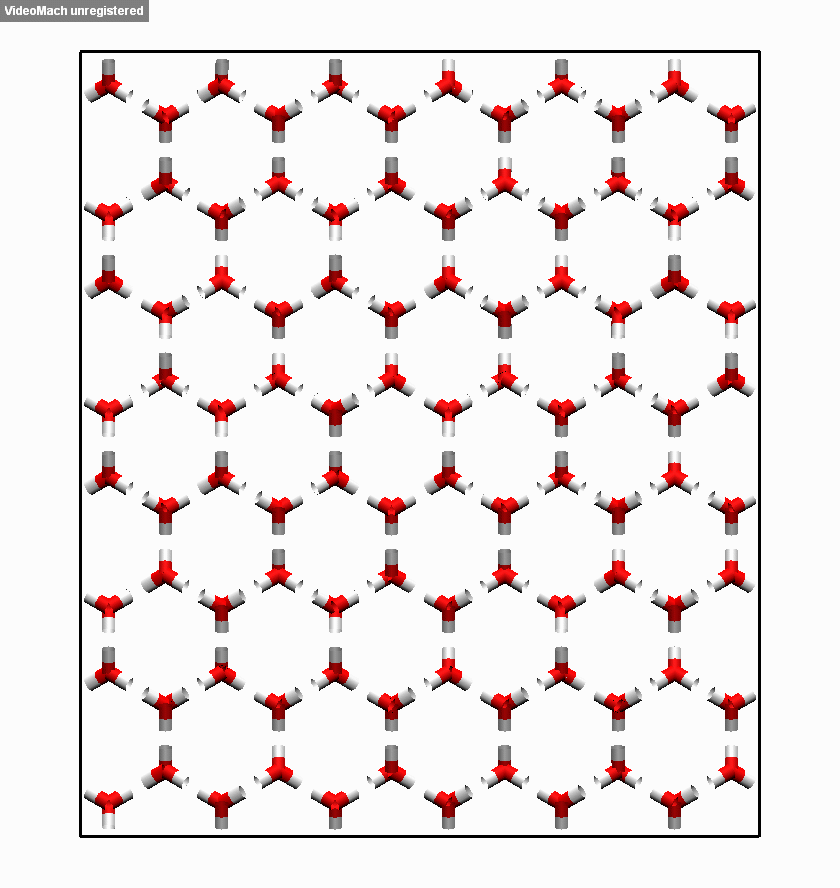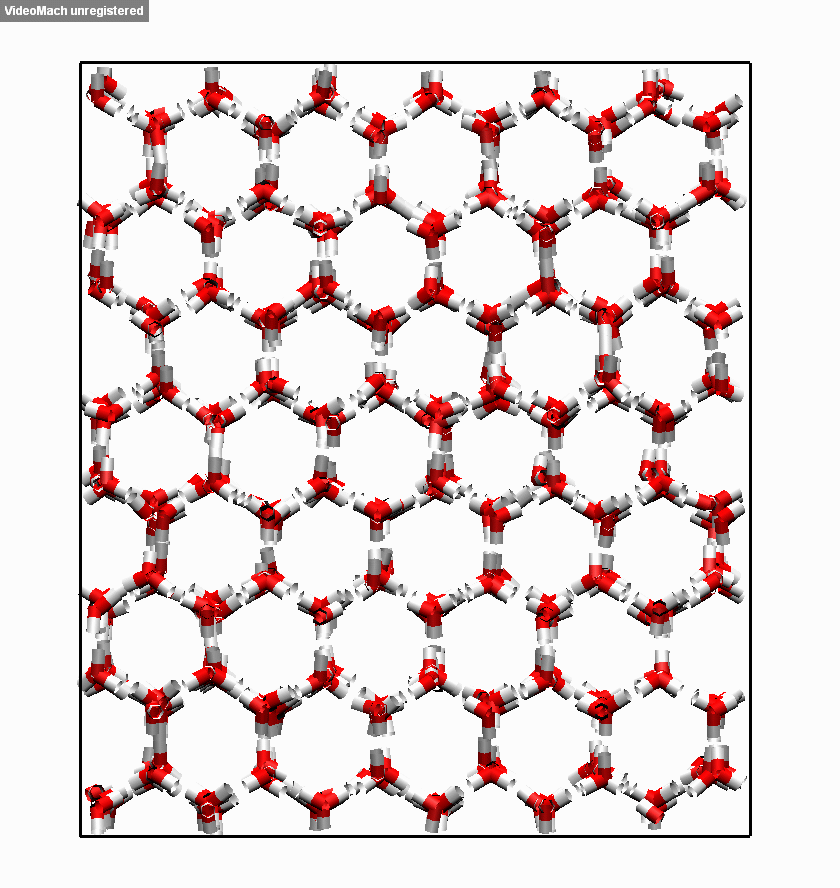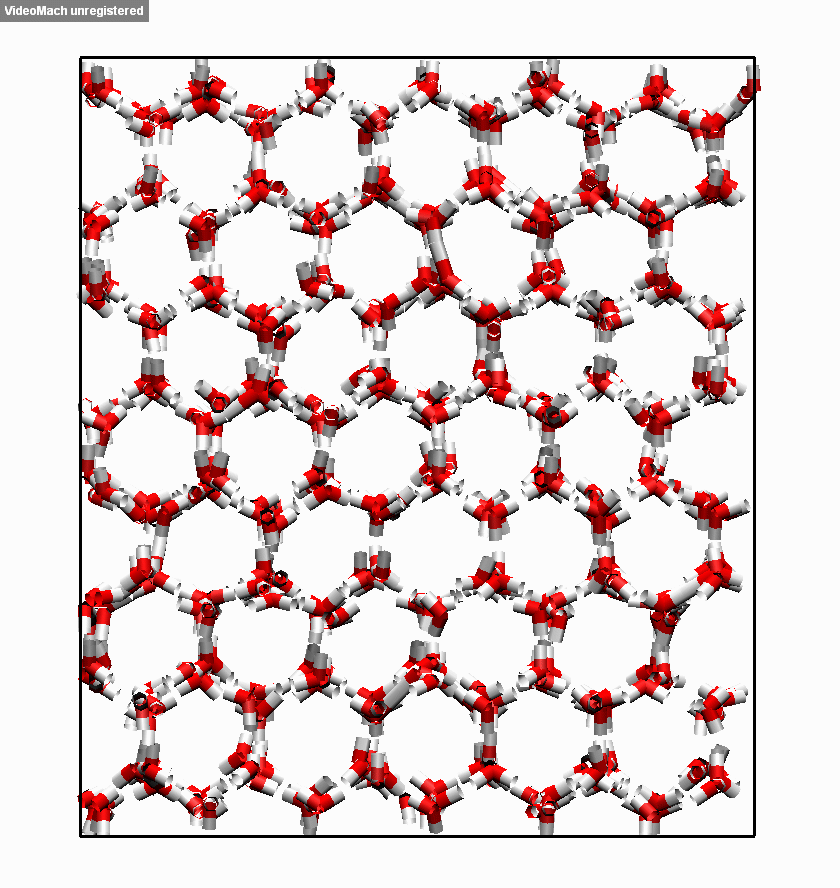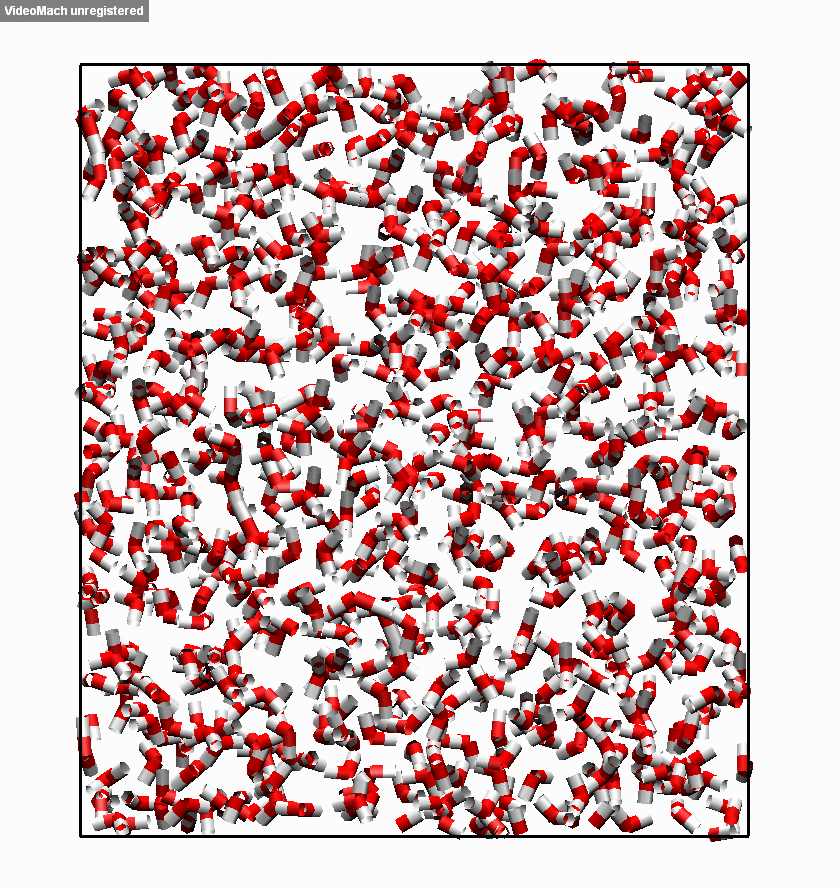
图3.3.5 冰融化过程
正辛醇和水相分离的MD模拟
正辛醇则是一种长链脂肪醇,分子中带有8个碳原子和一个氧原子。由于分子结构中没有明显的极性键或官能团,正辛醇分子整体带有较低的极性。因此,正辛醇与水分子之间的相互作用力较弱,而水分子与水分子之间的相互作用力比较强,因此正辛醇只能微溶于水,会发生相分离现象。本案例通过联合原子力场建立正辛醇分子模型,选择OPC3力场建立水分子模型,继而进行分子动力学模拟相分离现象。其中AuToFF支持联合原子力场建立模型,其中联合原子力场是指与碳原子直接键连的氢原子的相对原子质量被重叠到碳原子上,形成一个联合原子的整体,同时,其他原子对氢原子的相互作用也被叠加到联合原子上,从而可以大幅度降低力场的复杂性,减少势参数。在联合原子力场中,大多数有机分子的力点数约为原子数的1/3(平均为CH2),MD模拟的计算效率大约可以提高一个数量级。
力场辅助工具-AuToFF
确定起始构型
选择AuToFF-联合原子力场模块,首先通过2D分子建模建立正辛醇结构简式,点击“生成3D结构视图”按钮即可3D显示,力场选择TraPPE联合原子力场。
图3.4.1 确定正辛醇结构
选择力场
正辛醇采用联合原子力场-TraPPE力场,AuToFF程序会根据分子自动匹配相应的原子类型
图3.4.2 根据力场选择原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件(top文件)的相关力场参数(itp文件,包括LJ、键、键角参数,原子电荷)。此外生成拓扑文件可支持多款计算软件,包括:GROMACS、LAMMPS、AMBER、Moltemplate、OpenMM、TINKER、CHARMM。
图3.4.3 生成正辛醇的力场拓扑文件
同理,对于水分子的力场拓扑文件生成选择全原子力场模块,力场选择OPC3即可,其余步骤同上
图3.4.4 确定水分子的力场类型
模拟体系建模
构建体系
首先,创建模拟体系。通过Packmol软件,我们将正辛醇分子和水分子放入一个立方体的模拟盒子中。这个过程中立方体的盒子大小要略大于同等密度下混合体系所需要的体积,以保证有足够的空间使得溶剂分子能够随机的分布并且模拟可以快速平衡。将AuToFF创建并下载好每个组分的拓扑文件,然后把pdb文件拷贝到packmol文件夹,调用packmol程序生成模拟的盒子。Packmol输入文件model.inp如下:
tolerance 2.0
filetype pdb
add_box_sides 1.5
output model.pdb
structure Oct.pdb
number 500
inside cube 0. 0. 0. 80
end structure
structure h2o.pdb
number 4000
inside cube 0. 0. 0. 80
end structure
运行 packmol < model.inp 可生成model.pdb文件,该文件包含了正辛醇和水混合模拟体系中所有原子的坐标和原子间的连接信息。将得到的model.pdb导入到VMD显示如下
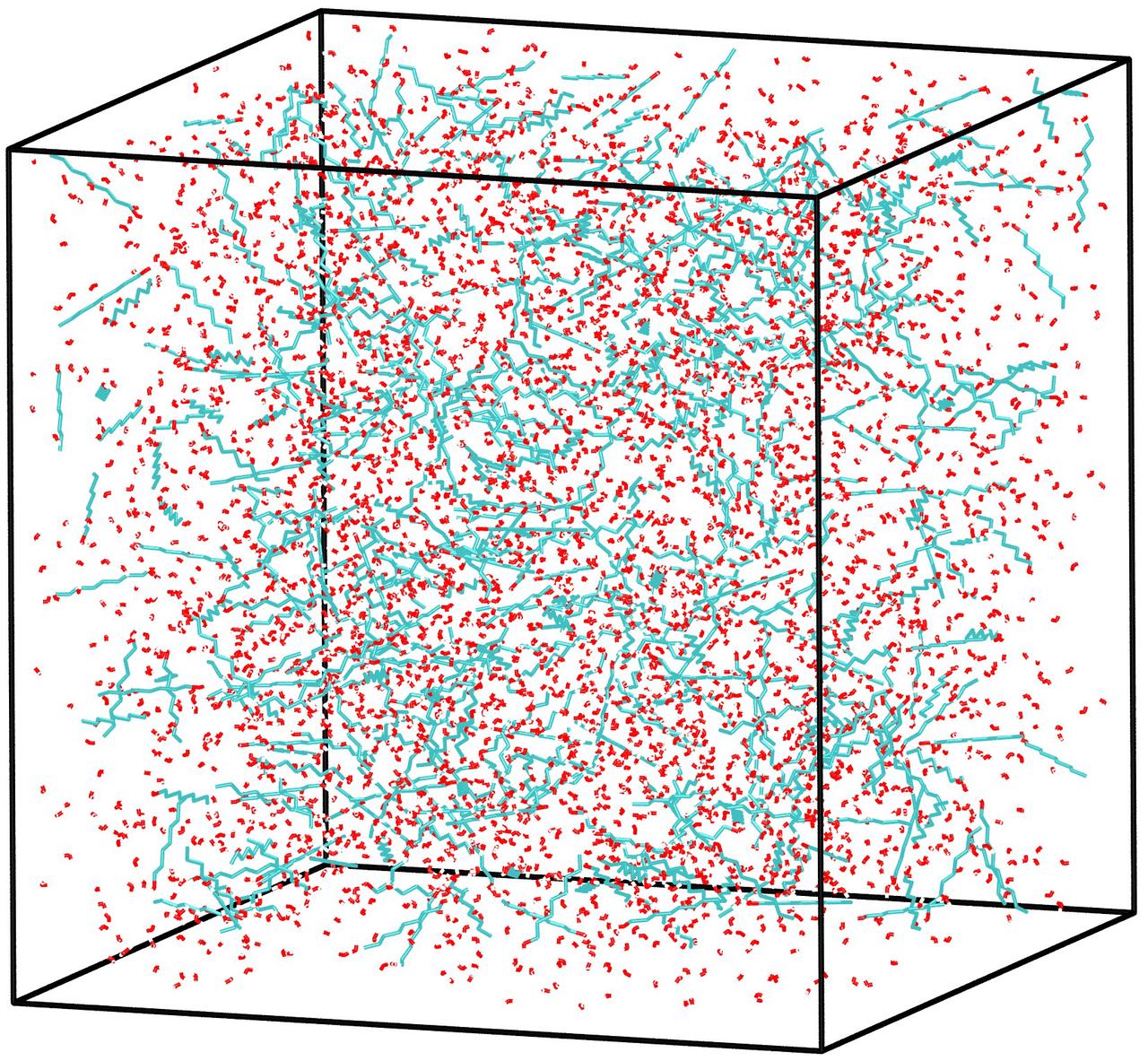
图3.4.5 正辛醇和水混合体系初始构型
构建拓扑文件
拓扑文件是gromacs运行模拟所必需的文件,它提供了模拟体系中所有分子的拓扑结构、力场文件的引用、约束力参数……;拓扑文件必须包含三个层次:
参数级别;这一部分包括了力场设定
分子定义级别:这一部分包含了一个或多个分子对应的.itp文件。实际上,.itp 文件可以看做是 .top 文件分子定义级别(针对每单个分子)单独拿出储存的信息,他们形成了一个嵌套式的引用关系
体系级别:只包含体系的特定信息
正辛醇和水混合模拟体系的top文件model.top如下:
[ defaults ]
1 3 yes 0 1
#include "Oct_ATP.itp"
#include "h2o_ATP.itp"
#include "Oct.itp"
#include "h2o.itp"
[ system ]
500Oct+4000h2o
[ molecules ]
Oct 500
h2o 4000
MD模拟
在模拟过程中,模拟步长设为2fs,积分算法选择速度Verlet算法。模拟体系的三个方向均考虑周期性,是体相的模拟。正辛醇-水混合体系的相分离过程模拟,采用梯度退火模拟。具体流程如下:等温等压系综下,模拟体系首先被缓慢加热到330 K,然后逐步将温度下降至目标温度298.15 K 。使用V-rescale控温,参考温度298.15 K, Berendsen控压, 参考压力为 1.01325 bar 。完整的GROMACS的mdp文件输入如下:
define =
integrator = md-vv-avek
dt = 0.002
nsteps = 2000000
comm-grps = system
energygrps =
;
nstxout = 0
nstvout = 0
nstfout = 0
nstlog = 500
nstenergy = 500
nstxout-compressed = 1000
compressed-x-grps = system
;
annealing = single
annealing_npoints = 3
annealing_time = 0 2000 4000
annealing_temp = 0 330 298.15
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = cut-off
rvdw = 1.0
DispCorr = EnerPres
;
Tcoupl = V-rescale
tau_t = 0.5
tc_grps = system
ref_t = 298.15
;
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 1
ref_p = 1.01325
compressibility = 8.5e-5
;
gen_vel = no
gen_temp = 298.15
gen_seed = -1
;
freezegrps =
freezedim =
constraints = hbonds
MD结果分析
MD过程的轨迹变化通过VMD作图如下,可以清晰的展现出水-正辛醇自发相分离现象,如下图所示,初始构型是混合体系,正辛醇和水均匀混合在一起,进行一段时间模拟后正辛醇和水相互分离开,并且正辛醇形成类似磷脂双层膜的结构,亲水的羟基头部朝水,而疏水的烃链尾巴朝内。

图3.4.6 正辛醇和水混合体系相分离模拟轨迹变化
黏度计算-AuToFF生成LAMMPS的力场拓扑data文件
黏度是影响电解液离子电导率的重要影响因素之一。黏度是电解液重要的基础物性,与电解液和电池的性能高度相关。能斯特–爱因斯坦方程揭示了黏度与粒子扩散的关联,表明黏度直接影响电解液中粒子的输运性质,从而影响电池的极化和倍率性能。此外,黏度会影响电解液对其他电池材料的润湿性,这也是在实际生产过程中必须考量的因素。下面利用 AuToFF 构建组分分子,并生成力场参数、电荷参数,继而使用 Packmol 建模获得体系中各原子坐标,再者 Moltemplate 补全体系拓扑信息、力场信息,并生成 LAMMPS 对应形式的力场拓扑data格式文件,最后采用LAMMPS进行非平衡态分子动力学方法(NEMD)方法模拟黏度性质。
力场辅助工具-AuToFF
确定起始构型
确定电解液溶剂EC分子结构,可选择点击AuToFF程序-全原子模块进行2D建模(如图1),点击“ 生成3d结构视图 ”按钮即可3D显示。

图3.5.1 创建EC分子结构
同上,依次建立EMC分子、 \(\ce{Li^+}\) 离子和 \(\ce{{PF_6}^-}\) 阴离子。此外 \(\ce{Li^+}\) 和 \(\ce{{PF_6}^-}\) 离子理应在离子液体模块建立。
选用适当力场和模拟软件
选择适当的力场是进行MD模拟的基础,可以快速地获得准确的模拟结果。针对溶剂分子EC和EMC选择GAFF2力场,而 \(\ce{Li^+}\) 离子选择CL&P力场,离子液体 \(\ce{{PF_6}^-}\) 选择OPLS-2009IL/0.8力场即可,确定原子类型。
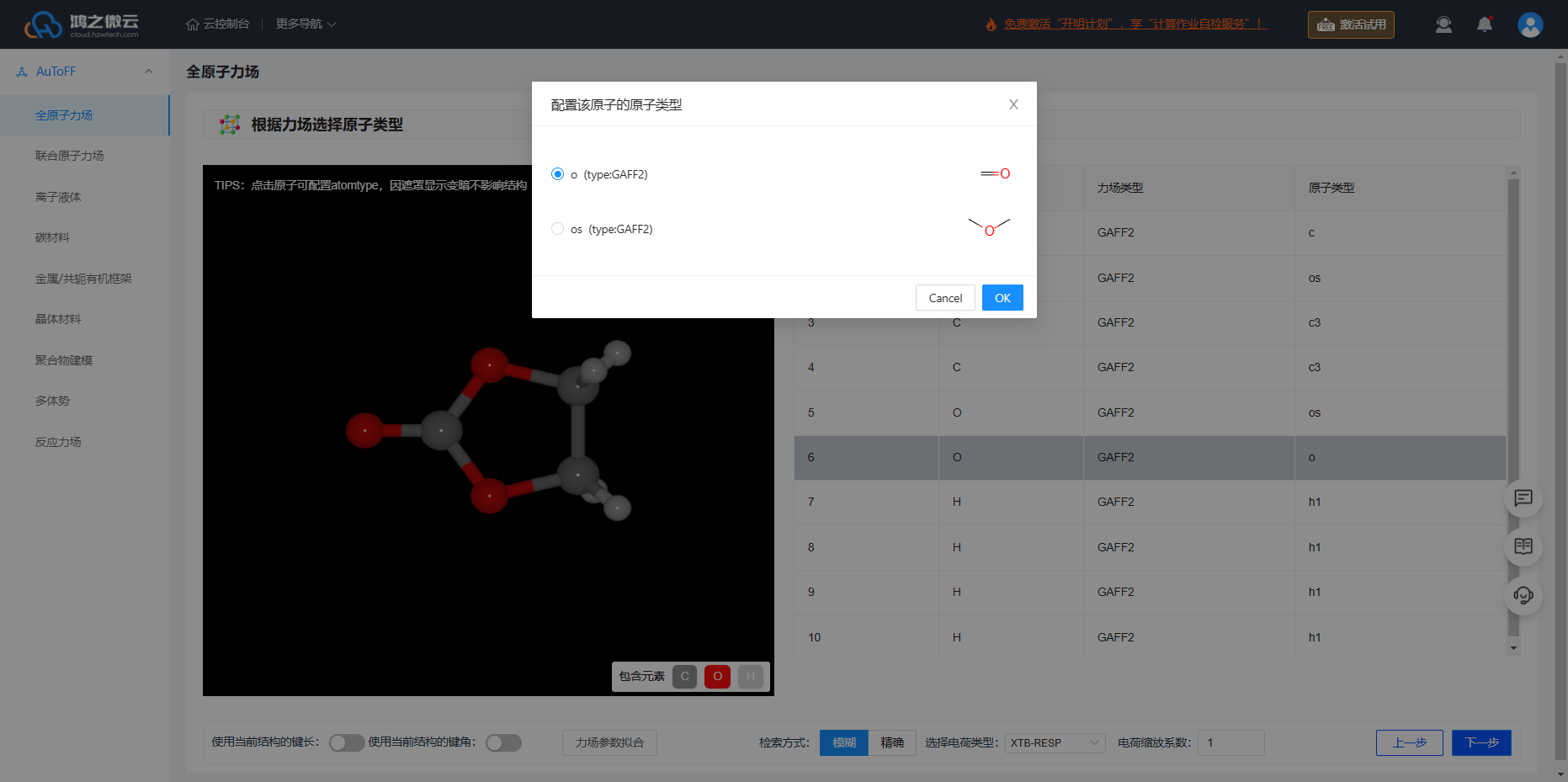
图3.5.2 根据力场选择原子类型
Note
点击结构视图中原子可进行配置原子类型
选择电荷类型:支持多种电荷计算方法,此处选择XTB-RESP,可以快速得到RESP近似电荷
生成拓扑文件
根据力场的选择即可生成拓扑文件的相关力场参数,包括LJ、键、键角、二面角参数、原子电荷,此外生成拓扑文件可支持多款计算软件。接下来,我们将结合Packmol和Moltemplate来实现复杂体系建模,并生成LAMMPS计算所需data力场拓扑文件。因此,选择Moltemplate生成该程序的输入文件。
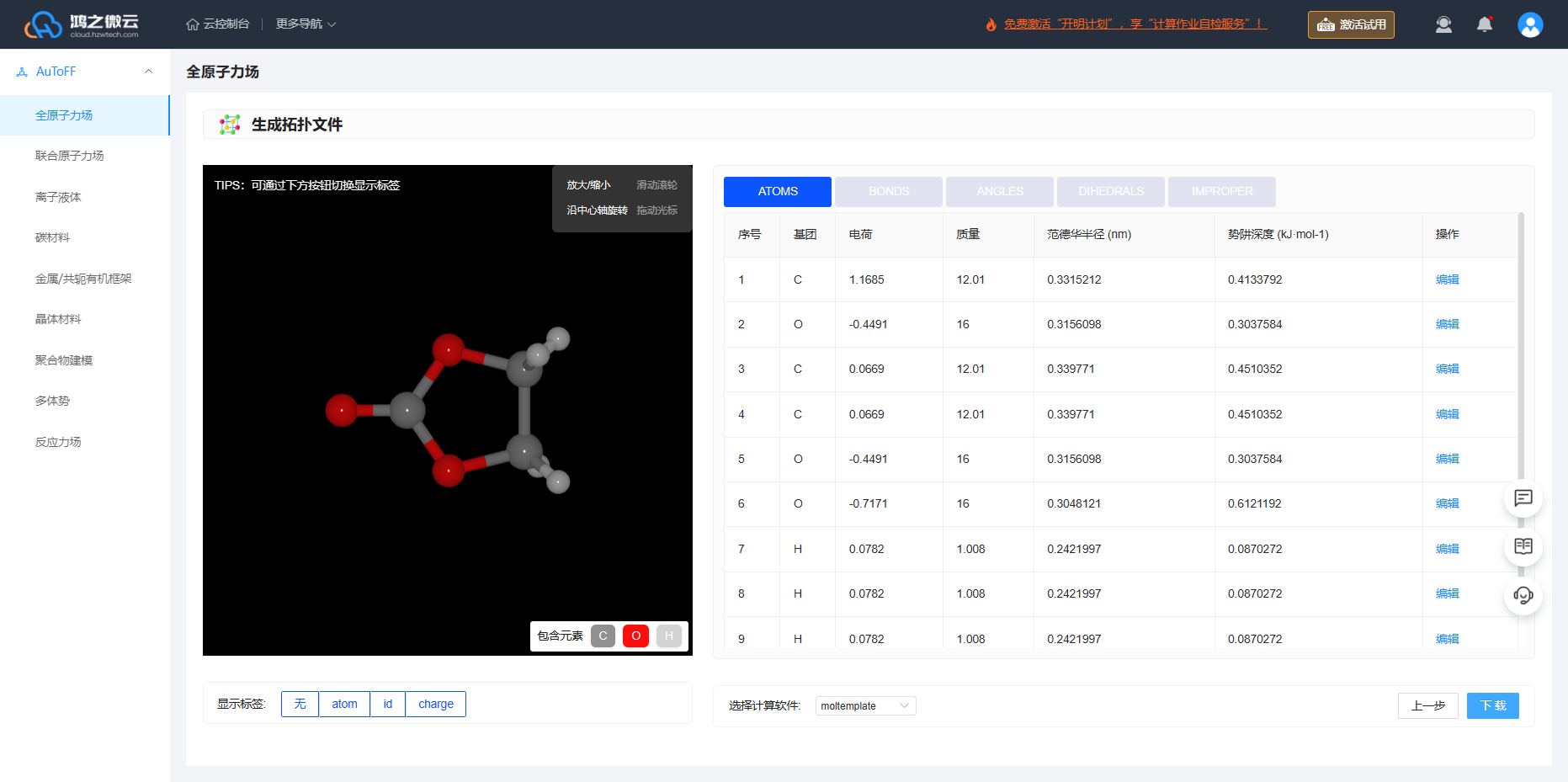
图3.5.3 生成拓扑文件
Note
点击下方显示标签按钮即可显示元素名称、原子ID、原子电荷。
用户也可通过 编辑 按钮进行自行修改原子电荷、力场参数信息。
由于 \(\ce{{PF_6}^-}\) 选择OPLS-2009IL/0.8力场,进行了电荷缩放,为保证体系呈电中性, \(\ce{Li^+}\) 离子电荷修改为0.8。
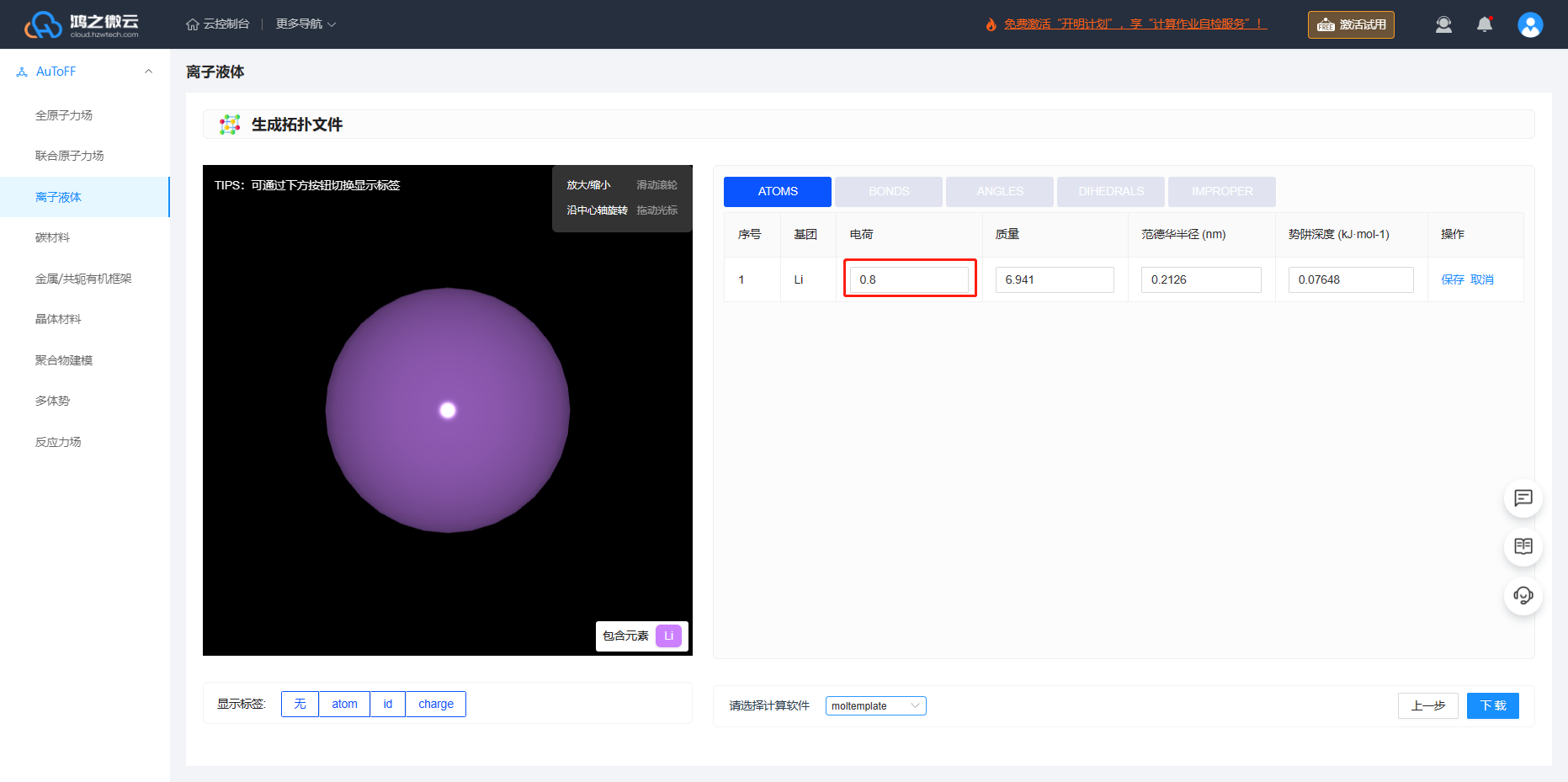
图3.5.4 锂离子电荷修改
一键下载的moltemplate压缩包中包含 各组分分子的pdb结构文件,Packmol的input文件,Moltemplate的lt输入文件
模拟体系建模
构建体系-Packmol
首先,创建模拟体系。通过Packmol软件,我们将电解液组成分子放入一个立方体的模拟盒子中。这个过程中立方体的盒子大小要略大于同等密度下电解液分子所需要的体积,以保证有足够的空间使得电解液分子能够随机的分布并且模拟可以快速平衡。将上步AuToFF创建并下载好每个组分的拓扑文件,然后把pdb文件拷贝到packmol文件夹,调用packmol程序生成模拟的盒子。Packmol输入文件model.inp如下:
tolerance 2.0
filetype pdb
add_box_sides 1.5
output model.pdb
structure EC.pdb
number 157
inside cube 0. 0. 0. 50
end structure
structure EMC.pdb
number 309
inside cube 0. 0. 0. 50
end structure
structure Li.pdb
number 45
inside cube 0. 0. 0. 50
end structure
structure PF6.pdb
number 45
inside cube 0. 0. 0. 50
end structure
运行 packmol < model.inp 可生成model.pdb文件,该文件包含了电解液模拟体系中所有原子的坐标,但缺少键、键角等拓扑结构信息。将得到的 model.pdb 可导入到VMD显示查看。
构建力场拓扑文件-Moltemplate
力场拓扑文件是运行MD模拟所必需的文件,接下来将利用Packmol生成的体系原子坐标文件,结合Moltemplate补全拓扑信息、力场信息等,并生成LAMMPS的data格式文件。其中前面利用 AuToFF-生成拓扑文件 一步中已生成了电解液模拟体系中各个组分的Moltemplate输入文件,下载链接 EC.lt EMC.lt Li.lt PF6.lt
电解液体系中包含上述四个组分,moltemplate输入文件system.lt如下:
import "EC.lt"
import "EMC.lt"
import "Li.lt"
import "PF6.lt"
ec = new EC [157]
emc = new EMC [309]
li = new Li [45]
pf6 = new PF6 [45]
write_once("Data Boundary") {
0.00000 50.00000 xlo xhi
0.00000 50.00000 ylo yhi
0.00000 50.00000 zlo zhi
Note
Moltemplate输入文件system.lt中各个组分顺序需与Packmol输入文件model.inp组分顺序保持一致。
运行 moltemplate.sh -pdb model.pdb system.lt 即可生成电解液体系 system.data 拓扑信息文件和system.in.settings system.in.settings 力场信息文件,该文件可在LAMMPS中直接使用。system.in.init文件涵盖了组分分子的力场函数类型,包括非键、键、角、二面角、赝扭曲势。
MD模拟-LAMMPS
借助分子动力学软件LAMMPS,利用非平衡态分子动力学方法计算体系黏度,即在非平衡态下对体系施加剪切,计算不同剪切速率下的稳态粘度,外推至零剪切速率下得到零切粘度。LAMMPS计算参数in.msd输入文件如下:
#-------------------------------------------------------------------------------------------------------------------#
variable temperature equal 298
variable timestep equal 1
variable Tdamp equal ${timestep}*100
variable Pdamp equal ${timestep}*1000
variable drag equal 0.7
variable tequ equal 1000
variable trun equal 1000000
variable srate equal 0.003
variable scaling equal 1e6/1e15
#-------------------------------------------------------------------------------------------------------------------#
units real
boundary p p p
atom_style full
pair_style lj/cut/coul/long 15.0
pair_modify mix arithmetic
special_bonds lj 0.0 0.0 0.5 coul 0.0 0.0 0.8333
kspace_style pppm 1.0e-5
bond_style harmonic
angle_style harmonic
dihedral_style fourier
improper_style cvff
read_data system.data #导入体系拓扑信息文件
include system.in.settings #导入体系力场信息文件
thermo 1000
timestep ${timestep}
#-------------------------------------------------------------------------------------------------------------------#
#minimize 1.0e-4 1.0e-6 100 1000
minimize 0.0 1.0e-8 1000 100000
fix 1 all nve/limit 0.1
fix 2 all langevin ${temperature} ${temperature} ${Tdamp} 123456 zero yes
run 1000
unfix 2
unfix 1
#-------------------------------------------------------------------------------------------------------------------#
fix npt all npt temp ${temperature} ${temperature} ${Tdamp} iso 0 0 ${Pdamp} drag ${drag}
run ${tequ}
unfix npt
write_data data.final
reset_timestep 0
#-------------------------------------------------------------------------------------------------------------------#
change_box all triclinic
kspace_style pppm 1.0e-5
fix deform all deform 1 xy erate ${srate} remap v
fix sllod all nvt/sllod temp ${temperature} ${temperature} ${Tdamp}
compute usual all temp
compute tilt all temp/deform
thermo_style custom step temp c_usual epair etotal press pxy
thermo_modify temp tilt
#--------------------------------------------------------------------------------------------------------#
fix rescale all temp/rescale 1 ${temperature} ${temperature} 1.0 1.0
run 10000
unfix rescale
run 10000
reset_timestep 0
#--------------------------------------------------------------------------------------------------------#
variable visc equal -pxy/(${srate})*${scaling}
fix vave all ave/time 10 100 1000 v_visc ave running start 500000
thermo_style custom step temp press pxy v_visc f_vave
thermo_modify temp tilt
run ${trun}
MD结果分析
黏度分析
分析LAMMPS输出文件log.lammps得到黏度性质,其中LAMMPS输入文件已进行结果预处理,输出f_vave数值已进行平均处理,直接读取最后一步的数据即可,即为稳态黏度数值。此外,还模拟了不同剪切速率下的稳态黏度,结果展示如下图:
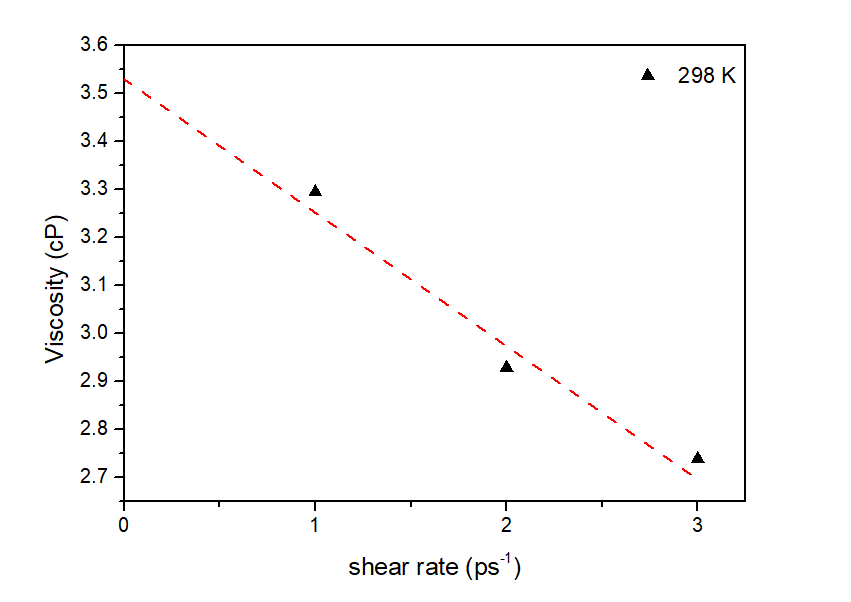
图3.5.5 不同剪切速率下的体系稳态粘度
沸石分子筛吸附 \(\ce{CH_4}\) 气体的饱和吸附曲线GCMC模拟
沸石是一类重要的多孔晶体材料,它们通过氧桥互相连接形成各种拓扑结构,普遍应用在煤化工、石油化工和精细化工等领域,对 \(\ce{CH_4}\) 分子有良好的吸附分离性能。因此,本案例我们将以ASV分子筛为例,利用AuToFF建模和生成力场参数文件,进而作为RASPA软件的输入文件进行巨正则蒙特卡罗模拟方法, 获得了沸石分子筛对 \(\ce{CH_4}\) 气体的吸附等温曲线。
力场辅助工具-AuToFF
确定起始构型
选择AuToFF-晶体材料模块,选择沸石 zeolites 文件夹- ASV.cif 完成结构建模,点击 生成3d结构视图 按钮即可3D显示,继而选择 TraPPE-zeo 力场开始进行力场参数分配。
图3.6.1 选择ASV晶体文件
由于 \(\ce{CH_4}\) 分子将采用联合原子力场,因此直接在联合原子力场模块界面进行模型搭建。
图3.6.2 创建 \(\ce{CH_4}\) 分子结构
根据力场选择原子类型
TraPPE-zeo是特定针对沸石骨架体系的力场。
图3.6.3 ASV晶体根据力场选择原子类型
对于 \(\ce{CH_4}\) 分子, 采用联合原子势能模型,即TraPPE-UA。
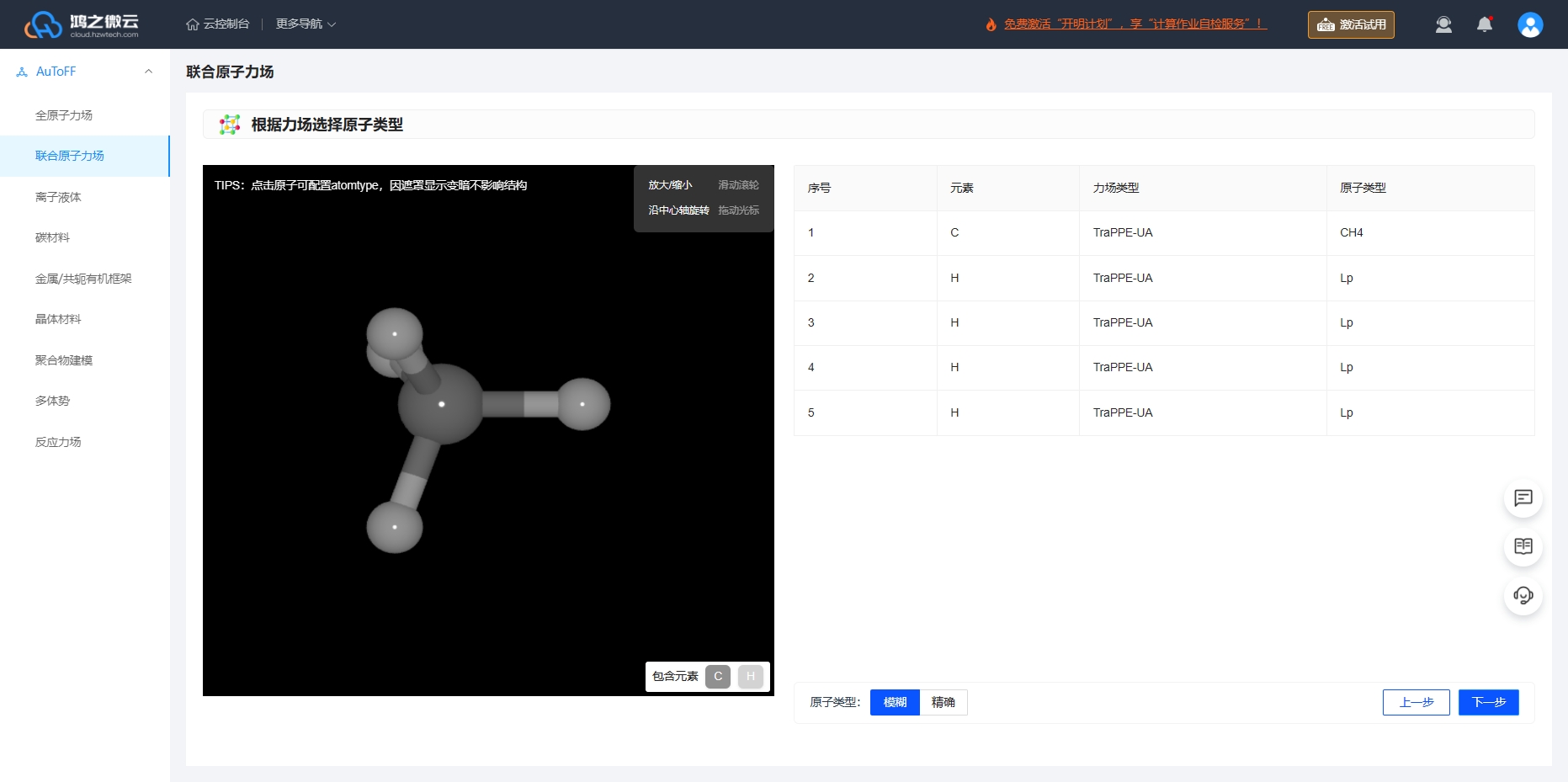
图3.6.4 \(\ce{CH_4}\) 分子根据力场选择原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件的相关力场参数,Lennard-Jones (LJ)势能函数和Coulomb势能函数共同描述吸附质分子之间以及客体分子与沸石骨架原子的非键相互作用。选择RASPA软件即可下载对应软件格式的力场输入文件和结构文件。
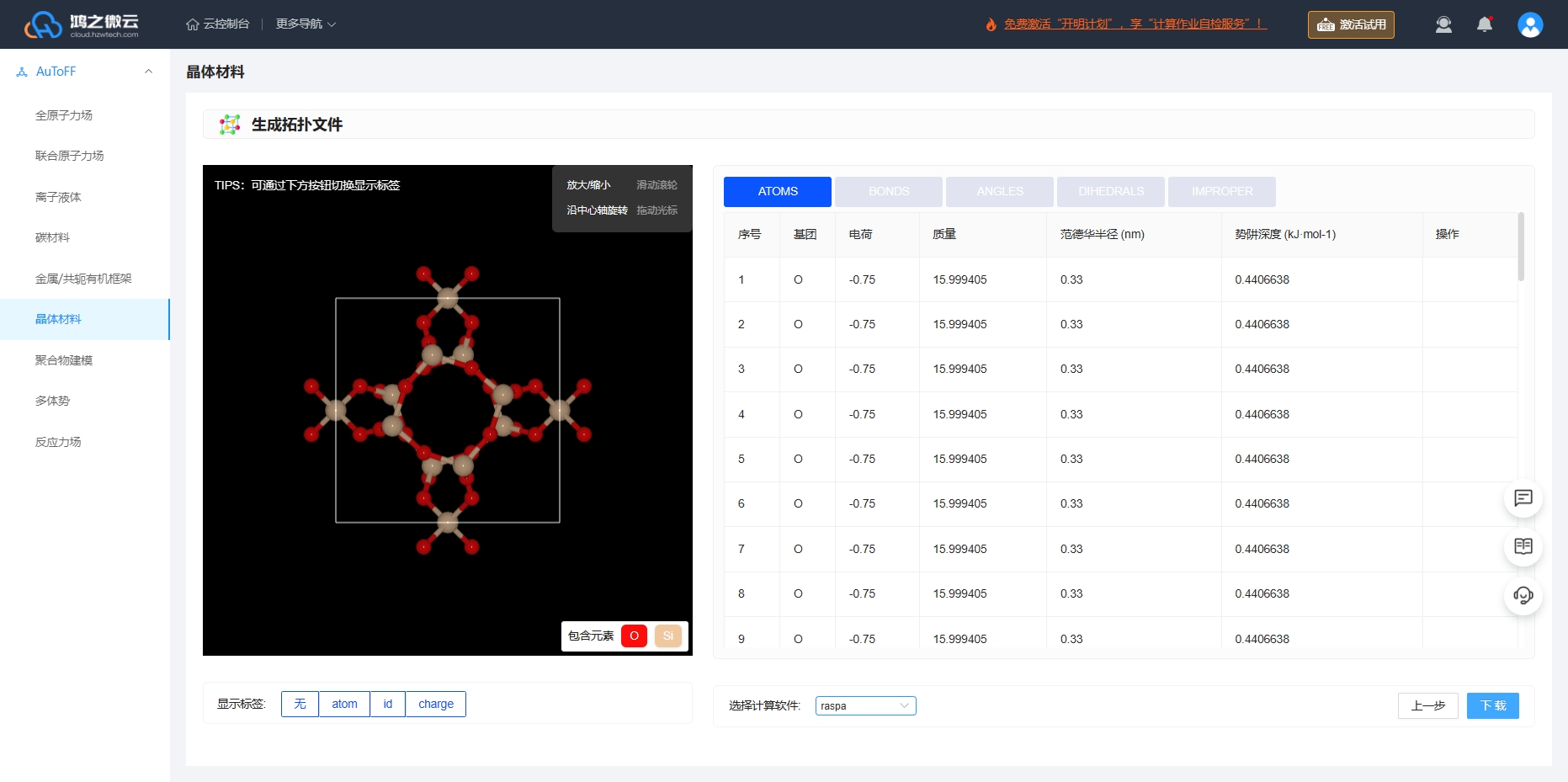
图3.6.5 生成ASV沸石分子筛晶体的拓扑文件
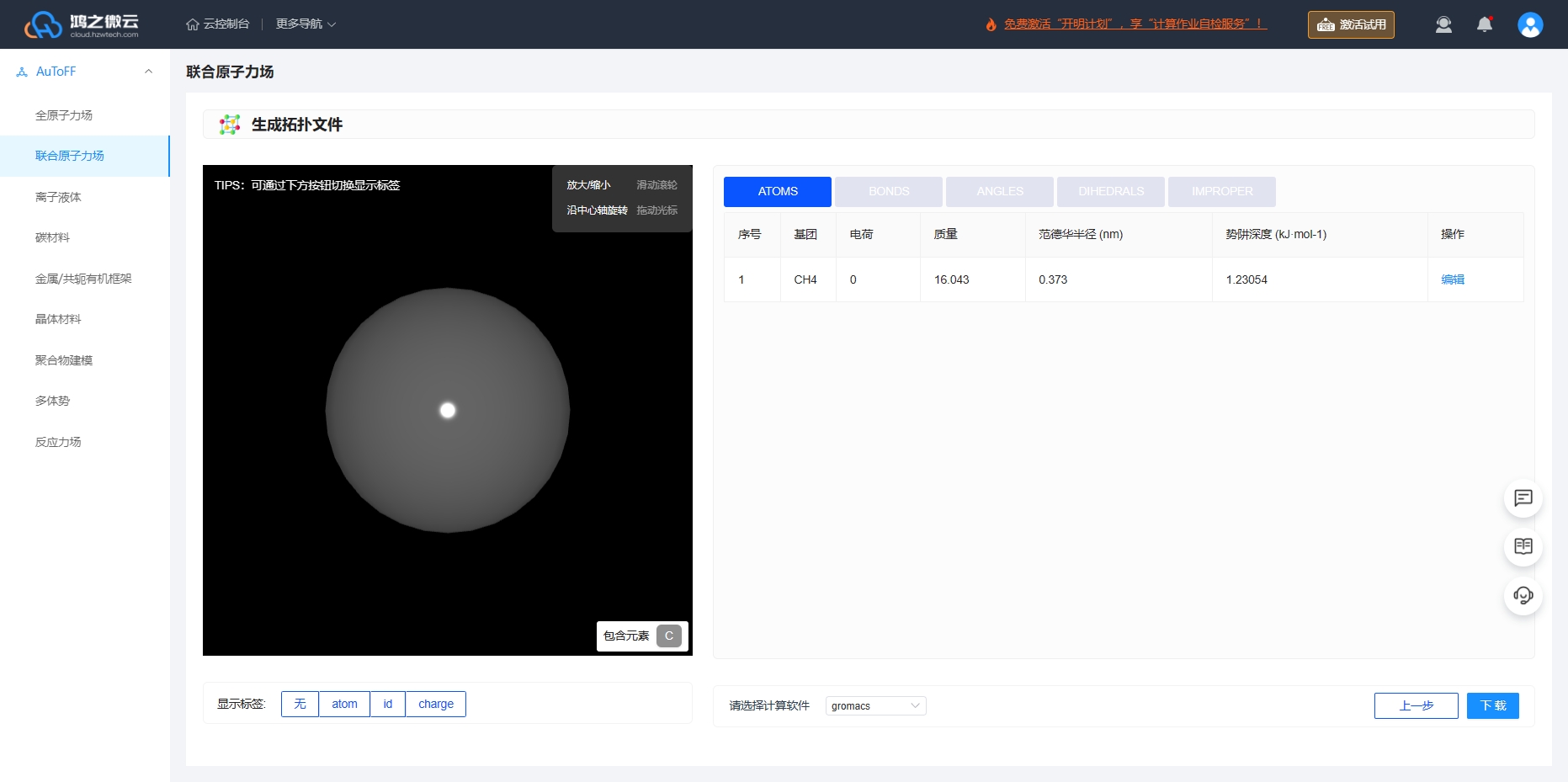
图3.6.6 生成 \(\ce{CH_4}\) 分子的拓扑文件
GCMC模拟-RASPA
采用GCMC模拟方法计算ASV沸石结构的甲烷吸附量,具体运行所需文件详见如下:
simulation.input 输入文件
包含模拟类型, 模拟的步数, 骨架名字, 晶胞数目, 使用的小分子, Monte-Carlo 行动 (move) 类型等控制词条。
SimulationType MonteCarlo #模拟方法为蒙特卡洛方法
NumberOfCycles 25000 #初始化结束后,用于生成平衡构象(采样)的循环次数。
NumberOfInitializationCycles 2000 #使用蒙特卡洛法初始化系统所使用的循环次数。
PrintEvery 1000 #每隔1000步进行输出打印
Forcefield local #选择当前目录的力场文件,此处直接利用AuToFF一键生成的力场文件
RemoveAtomNumberCodeFromLabel yes #在读取cif格式Framework信息时,将元素后面的序号都抹去,极大减少了计算量与分配力场参数的工作量
Framework 0
FrameworkName asv #定义多孔材料结构名称
UnitCells 3 3 2 #定义晶胞数量3*3*2
HeliumVoidFraction 0.21 #定义多孔材料的孔隙率,计算方法见案例7 RASPA计算孔隙率
ExternalTemperature 300.0
ExternalPressure 1e4
Component 0 MoleculeName ch4 #定义吸附质名称
MoleculeDefinition local
FugacityCoefficient 1.0
TranslationProbability 0.5 #平移概率
RegrowProbability 0.5 #重生概率
SwapProbability 1.0 #交换概率
CreateNumberOfMolecules 0 #GCMC模拟应始终为0
structure-name.cif 结构文件
多孔材料的结构文件,AuToFF中下载的压缩包中包含结构文件,ASV沸石骨架结构下载链接 asv.cif
pseudo_atoms.def 结构文件
列举使用的赝原子的信息,包括电荷和质量等。一般情况下赝原子代表一个原子,但也可能代表一个小基团 (比如 -CH3)。由于 CIF 文件会提供原子信息,因此在 CIF 中列举的原子并不需要在赝原子列表中进行规定,当读取 CIF 文件时原子信息将自动加入到该列表中。如果在赝原子中也提供了原子信息,那么该文件中的数据将被优先读取。 AuToFF分别下载 \(\ce{CH_4}\) 分子和ASV沸石的拓扑文件,两个文件夹中的pseudo_atoms.def进行整合,内容如下:
#number of pseudo atoms. Created by AutoFF
3
# type print as scat oxidation mass charge polarization B-factor radii connectivity anisotropic anisotropic-type tinker-type
zeo_Si yes Si Si 0 28.085499 1.500000 0.0 1.0 1.0 0 0.0 relative 0
zeo_OZ yes O O 0 15.999405 -0.750000 0.0 1.0 1.0 0 0.0 relative 0
Tra_CH4 yes CH4 CH4 0 16.043000 0.000000 0.0 1.0 1.0 0 0.0 relative 0
force_field_mxing_rules.def 力场文件
定义每个原子的势参数和混合规则
# general rule for shifted vs truncated. Created by AutoFF
shift
# general rule tail corrections
no
# number of defined interactions
3
# type interaction, parameters. IMPORTANT: define generic matches first
Si lennard-jones 21.999858 2.300000
O lennard-jones 52.999673 3.300000
Tra_CH4 lennard-jones 147.999944 3.730000
# general mixing rule for Lennard-Jones
Lorentz-Berthelot
Note
为了降低计算量,输入文件RemoveAtomNumberCodeFromLabel变量设置了yes参数,意味着在读取cif格式地Framework信息时,将元素后面的序号都删除,因此force_field_mxing_rules.def文件中原子类型仅需修改成Si,O。
Framework.def 文件
Framework.def存储骨架结构键, 键角, 二面角的伸缩扭转等参数 (非必须) ,AuToFF中下载的压缩包中包含该文件,下载链接 asv.def
molecules.def 分子文件
由于simulation.input输入文件定义MoleculeDefinition参数为local,需在该目录存放该分子结构信息文件,即 ch4.def
结果分析
输出文件即可查看计算模拟所得ASV沸石对 \(\ce{CH_4}\) 的吸附量,即0.0346091881 mol/kg。
Number of molecules:
====================
Component 0 [ch4]
-------------------------------------------------------------
Block[ 0] 13.81640 [-]
Block[ 1] 13.71180 [-]
Block[ 2] 13.44740 [-]
Block[ 3] 13.13800 [-]
Block[ 4] 13.26120 [-]
------------------------------------------------------------------------------
Average 13.4749600000 +/- 0.5158832364 [-]
Average loading absolute [molecules/unit cell] 0.7486088889 +/- 0.0286601798 [-]
Average loading absolute [mol/kg framework] 0.0346091881 +/- 0.0013249984 [-]
Average loading absolute [milligram/gram framework] 0.5552352044 +/- 0.0212569488 [-]
Average loading absolute [cm^3 (STP)/gr framework] 0.7757295023 +/- 0.0296984812 [-]
Average loading absolute [cm^3 (STP)/cm^3 framework] 1.4780989560 +/- 0.0565884035 [-]
Block[ 0] 13.81640 [-]
Block[ 1] 13.71180 [-]
Block[ 2] 13.44740 [-]
Block[ 3] 13.13800 [-]
Block[ 4] 13.26120 [-]
------------------------------------------------------------------------------
Average 12.4749600000 +/- 0.5158832364 [-]
Average loading excess [molecules/unit cell] 0.6930533333 +/- 0.0286601798 [-]
Average loading excess [mol/kg framework] 0.0320407806 +/- 0.0013249984 [-]
Average loading excess [milligram/gram framework] 0.5140302432 +/- 0.0212569488 [-]
Average loading excess [cm^3 (STP)/gr framework] 0.7181612793 +/- 0.0296984812 [-]
Average loading excess [cm^3 (STP)/cm^3 framework] 1.3684066856 +/- 0.0565884035 [-]
同理,继续通过GCMC方法模拟了超微孔沸石材料 ASV 在300 K下、压力范围为 0—100 kPa 下的 \(\ce{CH_4}\) 单组份吸附性能,吸附曲线如下:
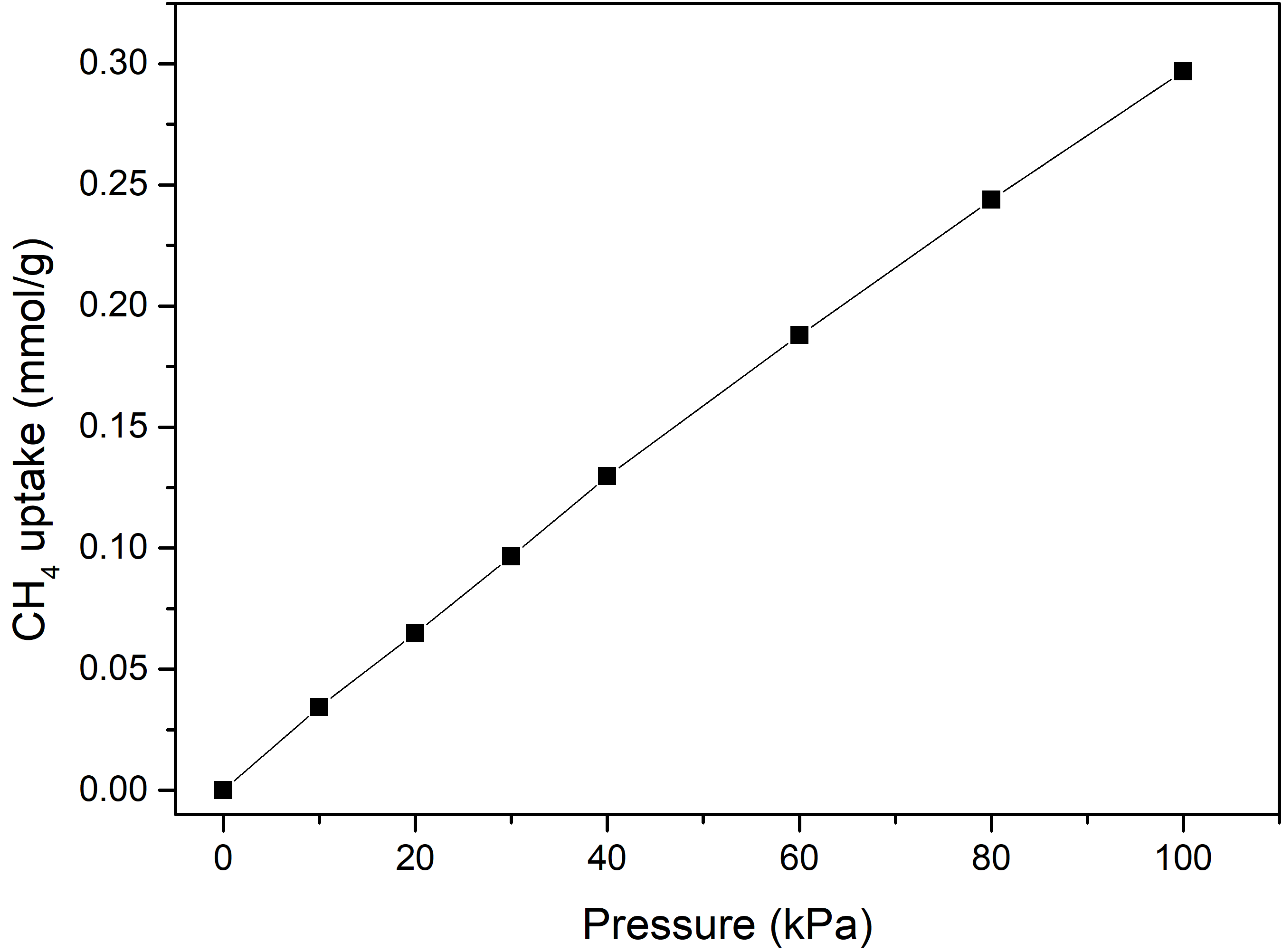
图3.6.7 沸石对 \(\ce{CH_4}\) 的吸附量
RASPA计算孔隙率
孔隙率是描述分子筛内部空隙含量的重要参数。沸石分子筛具有非常高的孔隙率,大的内表面积和约50%体积的空腔。在高温活化沸石失水后,在晶体内部形成大量具有均匀孔径的孔,其具有强吸附能力并且可以有效地将直径小于其孔径的分子吸入孔中,并且大于孔径的分子被阻挡在孔外,从而分离出不同大小的分子。可利用RASPA计算孔隙率,以氦气作为探针。具体操作详见如下。
力场辅助工具-AuToFF
确定起始构型
选择AuToFF-晶体材料模块,选择沸石 zeolites 文件夹- ASV.cif 完成结构建模,点击 生成3d结构视图 按钮即可3D显示,继而选择 TraPPE-zeo 力场开始进行力场参数分配。
图3.7.1 选择ASV晶体文件
根据力场选择原子类型
TraPPE-zeo是特定针对沸石骨架体系的力场。
图3.7.2 ASV晶体根据力场选择原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件的相关力场参数,Lennard-Jones (LJ)势能函数和Coulomb势能函数共同描述吸附质分子之间以及客体分子与沸石骨架原子的非键相互作用。选择RASPA软件即可下载对应软件格式的力场输入文件和结构文件。
图3.7.3 生成ASV沸石分子筛晶体的拓扑文件
MC模拟-RASPA
采用MC模拟方法以氦气作为探针,计算ASV沸石结构的孔隙率,具体运行所需文件详见如下:
simulation.input 输入文件
包含模拟类型, 模拟的步数, 骨架名字, 晶胞数目, 使用的小分子, Monte-Carlo 行动 (move) 类型等控制词条。
SimulationType MonteCarlo
NumberOfCycles 10000
PrintEvery 1000
PrintPropertiesEvery 1000
Forcefield local
RemoveAtomNumberCodeFromLabel yes
Framework 0
FrameworkName asv
UnitCells 3 3 2
ExternalTemperature 298.0
Component 0 MoleculeName helium
MoleculeDefinition local
WidomProbability 1.0
CreateNumberOfMolecules 0
structure-name.cif 结构文件
多孔材料的结构文件,AuToFF中下载的压缩包中包含结构文件,ASV沸石骨架结构下载链接 asv.cif
pseudo_atoms.def 结构文件
列举使用的赝原子的信息,包括电荷和质量等。一般情况下赝原子代表一个原子,但也可能代表一个小基团 (比如 -CH3)。由于 CIF 文件会提供原子信息,因此在 CIF 中列举的原子并不需要在赝原子列表中进行规定,当读取 CIF 文件时原子信息将自动加入到该列表中。如果在赝原子中也提供了原子信息,那么该文件中的数据将被优先读取。 AuToFF下载ASV沸石的拓扑文件,对ASV晶体和氦气分子的pseudo_atoms.def进行整合,内容如下:
#number of pseudo atoms. Created by AutoFF
3
# type print as scat oxidation mass charge polarization B-factor radii connectivity anisotropic anisotropic-type tinker-type
zeo_Si yes Si Si 0 28.085499 1.500000 0.0 1.0 1.0 0 0.0 relative 0
zeo_OZ yes O O 0 15.999405 -0.750000 0.0 1.0 1.0 0 0.0 relative 0
He yes He He 0 4.002602 0.0 0.0 1.0 1.0 0 0.0 relative 0
Note
pseudo_atoms.def中的He参数详见RASPA2-master/forcefield/GarciaPerez2006/pseudo_atoms.def
force_field_mxing_rules.def 力场文件
定义每个原子的势参数和混合规则
# general rule for shifted vs truncated. Created by AutoFF
shift
# general rule tail corrections
no
# number of defined interactions
3
# type interaction, parameters. IMPORTANT: define generic matches first
Si lennard-jones 21.999858 2.300000
O lennard-jones 52.999673 3.300000
He lennard-jones 10.9 2.64
# general mixing rule for Lennard-Jones
Lorentz-Berthelot
Note
为了降低计算量,输入文件设置了RemoveAtomNumberCodeFromLabel参数,因此force_field_mxing_rules.def文件中原子类型仅需修改成Si,O
force_field_mxing_rules.def中的He参数详见RASPA2-master/forcefield/GarciaPerez2006/force_field_mixing_rules.def
Framework.def 文件
Framework.def存储骨架结构键, 键角, 二面角的伸缩扭转等参数 (非必须) ,AuToFF中下载的压缩包中包含该文件,下载链接 asv.def
molecules.def 分子文件
由于simulation.input输入文件定义MoleculeDefinition参数为local,需在该目录存放该分子结构信息文件,即 helium.def
结果分析
输出文件即可查看计算模拟所得ASV沸石分子筛的孔隙率,即为0.21
Average Widom Rosenbluth factor:
================================
Block[ 0] 0.213241 [-]
Block[ 1] 0.212941 [-]
Block[ 2] 0.211396 [-]
Block[ 3] 0.21113 [-]
Block[ 4] 0.210937 [-]
------------------------------------------------------------------------------
[helium] Average Widom Rosenbluth-weight: 0.211929 +/- 0.001929 [-]
沸石分子筛的甲烷/氢气吸附选择性模拟
分子筛作为一种重要的材料,在气体处理与分离方面具有广泛的应用潜力。气体处理与分离是工业、环境和能源领域中非常重要的研究领域。其中,重要的应用包括天然气加工、石油化工、空气净化、环境污染治理等。在这些应用中,气体处理与分离面临着许多挑战,例如:
气体混合物的复杂性:气体混合物通常由多种组分组成,包括不同的气体分子、杂质、水蒸汽等。这些组分之间的相互作用和竞争性吸附会影响到分离的效率和选择性。
目标组分的低浓度:在一些应用中,需要从低浓度的气体混合物中分离出目标组分。例如,从二氧化碳含量较低的气流中提取二氧化碳,需要具有高效的选择性吸附材料。
处理规模的不同:气体处理与分离的规模从小到大,从个人使用到工业生产都有涉及。因此,需要针对不同规模的处理应用,开发不同的材料和设备。
能源消耗:气体处理与分离通常需要大量的能源投入,例如吸附、膜分离等过程需要在高压下进行,消耗大量的电力和气体。
以上是气体处理与分离领域面临的挑战,解决这些挑战需要不断地发展新材料和技术,并结合实际应用需求进行研究。分子筛在气体处理与分离领域具有广泛的应用研究领域。以下是其中一些关键的应用研究领域:
气体吸附分离:分子筛可以通过选择性吸附不同气体组分来实现气体的分离。例如,通过调节分子筛的孔径和表面性质,可以实现二氧化碳的捕获与封存、甲烷的选择性吸附等。此外,分子筛还可用于空气中的有害气体去除,如去除甲醛、苯等有机污染物。
催化氧化:分子筛被广泛应用于催化反应中,其中包括气体处理与分离领域。例如,在汽车尾气处理中,分子筛可以作为催化剂的载体,通过催化氧化将有害气体转化为无害物质,如将一氧化碳转化为二氧化碳。
脱硫脱氮:分子筛可以用于脱除燃烧废气中的硫氧化物和氮氧化物。通过选择性吸附和催化反应,分子筛可以将硫氧化物和氮氧化物转化为无害物质,从而减少对环境的影响。
气体传感:分子筛可以用于气体传感器的制备。通过选择性吸附特定气体,分子筛可以实现对目标气体的高灵敏度检测。这在环境监测、生命安全等领域具有重要应用价值。
这些应用研究领域也充分说明了分子筛在气体处理与分离中的重要性和潜力。通过不断筛选和开发新材料、调节操作条件,可以进一步提高分子筛的性能,拓宽其应用范围,并为解决气体处理与分离问题提供有效的解决方案。
分子筛在气体处理与分离方面的评价指标主要包括以下几个方面:
吸附容量:表示吸附剂单位质量或体积对目标组分吸附的能力。通常以吸附剂饱和时目标组分的吸附量来表示。
选择性系数:表示吸附剂在混合气流中选择性地吸附某一组分相对于其他组分的能力。通常定义为目标组分和杂质组分吸附量之比。
分离因子:表示吸附剂选择性吸附两种组分时,两种组分在吸附剂上的浓度比值与初始混合气流中的浓度比值的比值。分离因子越大,说明分离效果越好。
催化活性:表示催化剂对反应物转化的能力。通常定义为反应物转化率与时间的比值。
通过这些评价指标的研究,可以深入了解分子筛的性能和应用特点,并为进一步拓展分子筛在气体处理与分离领域的应用提供支持和指导。
下例我们通过GCMC方法着重探究了ASV沸石对 \(\ce{CH_4}\) / \(\ce{H_2}\) 的二元混合组份吸附,计算不同组分的混合气体体系中对 \(\ce{CH_4}\) 气体分子的选择性系数。
力场辅助工具-AuToFF
确定起始构型
选择AuToFF-晶体材料模块,选择沸石 zeolites 文件夹- ASV.cif 完成结构建模,点击 生成3d结构视图 按钮即可3D显示,继而选择 TraPPE-zeo 力场开始进行力场参数分配。
图3.8.1 选择ASV晶体文件
由于 \(\ce{CH_4}\) 分子将采用联合原子力场,因此直接在联合原子力场模块界面进行模型搭建。
图3.8.2 创建 \(\ce{CH_4}\) 分子结构
根据力场选择原子类型
TraPPE-zeo是特定针对沸石骨架体系的力场。
图3.8.3 ASV晶体根据力场选择原子类型
对于 \(\ce{CH_4}\) 分子, 采用联合原子势能模型,即TraPPE-UA。
图3.8.4 \(\ce{CH_4}\) 分子根据力场选择原子类型
生成拓扑文件
根据力场的选择即可生成拓扑文件的相关力场参数,Lennard-Jones (LJ)势能函数和Coulomb势能函数共同描述吸附质分子之间以及客体分子与沸石骨架原子的非键相互作用。选择RASPA软件即可下载对应软件格式的力场输入文件和结构文件。
图3.8.5 生成ASV沸石分子筛晶体的拓扑文件
图3.8.6 生成 \(\ce{CH_4}\) 分子的拓扑文件
GCMC模拟-RASPA
GCMC模拟了温度为 300 K 时沸石分子筛ASV晶体在 \(\ce{CH_4}\) / \(\ce{H_2}\) 混合体系中对 \(\ce{CH_4}\) 气体分子的吸附选择性能,具体运行所需文件详见如下:
simulation.input 输入文件
包含模拟类型, 模拟的步数, 骨架名字, 晶胞数目, 使用的小分子, Monte-Carlo 行动 (move) 类型等控制词条。
SimulationType MonteCarlo
NumberOfCycles 25000
NumberOfInitializationCycles 2000
PrintEvery 1000
#ContinueAfterCrash no
#WriteBinaryRestartFileEvery 2000
Forcefield local
RemoveAtomNumberCodeFromLabel yes
Framework 0
FrameworkName asv
UnitCells 3 3 2
HeliumVoidFraction 0.21
ExternalTemperature 300.0
ExternalPressure 1e5
Component 0 MoleculeName ch4
MoleculeDefinition local
MolFraction 0.5 #摩尔分数
FugacityCoefficient 1.0
TranslationProbability 0.5 #平移概率
RegrowProbability 0.5 #重生概率
IdentityChangeProbability 1.0 #改变身份概率
NumberOfIdentityChanges 2 #身份改变次数
IdentityChangesList 0 1 #身份互换组分列表
SwapProbability 1.0 #交换概率
CreateNumberOfMolecules 0 #交换概率
Component 1 MoleculeName H2
MoleculeDefinition local
MolFraction 0.5
FugacityCoefficient 1.0
TranslationProbability 0.5
RegrowProbability 0.5
IdentityChangeProbability 1.0
NumberOfIdentityChanges 2
IdentityChangesList 0 1
SwapProbability 1.0
CreateNumberOfMolecules 0
structure-name.cif 结构文件
多孔材料的结构文件,AuToFF中下载的压缩包中包含结构文件,ASV沸石骨架结构下载链接 asv.cif
pseudo_atoms.def 结构文件
列举使用的赝原子的信息,包括电荷和质量等。一般情况下赝原子代表一个原子,但也可能代表一个小基团 (比如 -CH3)。由于 CIF 文件会提供原子信息,因此在 CIF 中列举的原子并不需要在赝原子列表中进行规定,当读取 CIF 文件时原子信息将自动加入到该列表中。如果在赝原子中也提供了原子信息,那么该文件中的数据将被优先读取。 AuToFF分别下载 \(\ce{CH_4}\) 分子和ASV沸石的拓扑文件,两个文件夹中的pseudo_atoms.def以及 \(\ce{H_2}\) 的赝势原子参数进行整合,内容如下:
#number of pseudo atoms. Created by AutoFF
5
# type print as scat oxidation mass charge polarization B-factor radii connectivity anisotropic anisotropic-type tinker-type
zeo_Si yes Si Si 0 28.085499 1.500000 0.0 1.0 1.0 0 0.0 relative 0
zeo_OZ yes O O 0 15.999405 -0.750000 0.0 1.0 1.0 0 0.0 relative 0
Tra_CH4 yes CH4 CH4 0 16.043000 0.000000 0.0 1.0 1.0 0 0.0 relative 0
H_h2 yes H H 0 1.00794 0.468 0.0 1.0 0.7 0 0 relative 0
H_com no H H 0 0.0 -0.936 0.0 1.0 0.7 0 0 relative 0
Note
混合体系中 \(\ce{H_2}\) 分子采用 Darkrim 和 Levesque 开发的三点模型, 在双原子分子的质心 (center of mass, COM) 引入点电荷来重现实验中分子的四极矩现象。
pseudo_atoms.def中的H相关参数详见RASPA2-master/forcefield/GenericZeolites/pseudo_atoms.def
force_field_mxing_rules.def 力场文件
定义每个原子的势参数和混合规则
# general rule for shifted vs truncated. Created by AutoFF
shift
# general rule tail corrections
no
# number of defined interactions
6
# type interaction, parameters. IMPORTANT: define generic matches first
Si lennard-jones 21.999858 2.300000
O lennard-jones 52.999673 3.300000
Tra_CH4 lennard-jones 147.999944 3.730000
H_h2 lennard-jones 36.7 2.958
H_com none
H_h2 lennard-jones 36.7 2.958
# general mixing rule for Lennard-Jones
Lorentz-Berthelot
Note
为了降低计算量,输入文件RemoveAtomNumberCodeFromLabel变量设置了yes参数,意味着在读取cif格式地Framework信息时,将元素后面的序号都删除,因此force_field_mxing_rules.def文件中原子类型仅需修改成Si,O
force_field_mxing_rules.def中的H参数详见RASPA2-master/forcefield/GenericZeolites/force_field_mixing_rules.def
Framework.def 文件
Framework.def存储骨架结构键, 键角, 二面角的伸缩扭转等参数 (非必须) ,AuToFF中下载的压缩包中包含该文件,下载链接 asv.def
molecules.def 分子文件
由于simulation.input输入文件定义MoleculeDefinition参数为local,需在该目录存放该分子结构信息文件,即 H2.def 、 ch4.def
Note
H2.def详见RASPA2-master/molecules/TraPPE/H2.def
结果分析
在模拟 \(\ce{CH_4}\) / \(\ce{H_2}\) 分离过程中, 二元混合组份的吸附选择性系数计算公式如下:
300 K下、100 kPa时,超微孔沸石材料ASV在组分不同的 \(\ce{CH_4}\) / \(\ce{H_2}\) 混合体系中对 \(\ce{CH_4}\) 组份吸附选择性能模拟,结果如下:
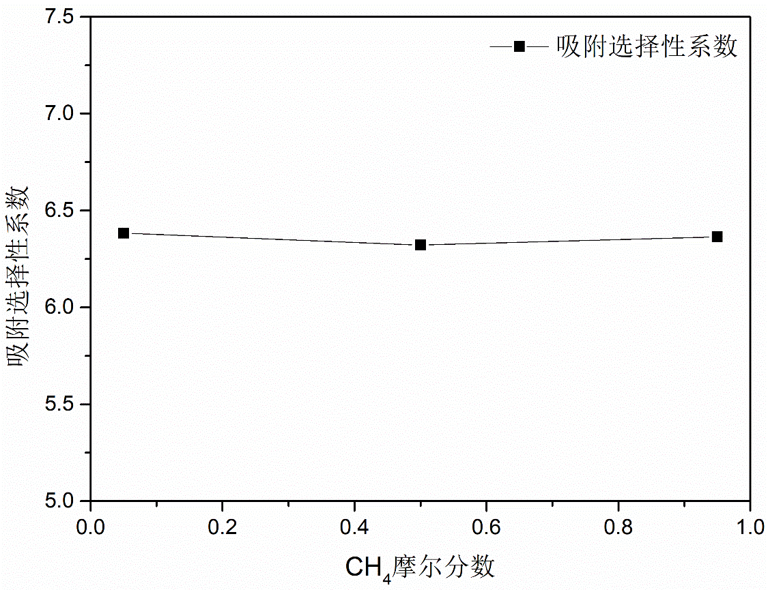
图3.8.7 吸附质分子进料比对沸石ASV的 \(\ce{CH_4}\) 选择性的影响
结果可见,吸附质分子的进料比对甲烷分子的分离性能影响比较小, 这也表现出ASV材料在不同环境下具有筛选甲烷分子的优良潜质。
300 K下时,模拟体系不同体相压力下,沸石ASV在等摩尔 \(\ce{CH_4}\) / \(\ce{H_2}\) 混合体系中对 \(\ce{CH_4}\) 的吸附选择性,结果如下:
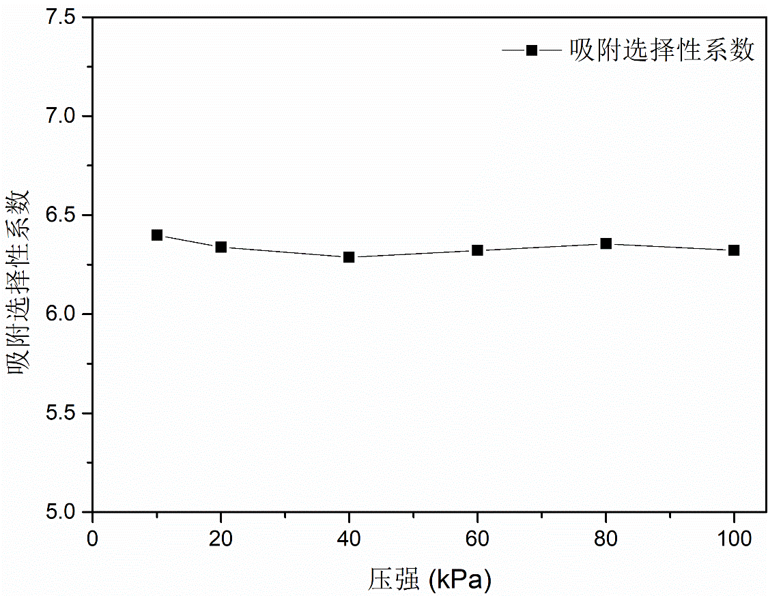
图3.8.8 压力对沸石ASV的 \(\ce{CH_4}\) 选择性的影响
由此可见,在 \(\ce{CH_4}\) / \(\ce{H_2}\) 系统中沸石ASV对 \(\ce{CH_4}\) 的吸附选择性几乎与体相压力的变化无关。这说明沸石的结构特征直接决定了 \(\ce{H_2}\) 气体分子的吸附选择性。