快速入门
本章将介绍AuToFF的各种功能,并针对具体的功能给出基本的使用说明。
全原子力场
全原子力场 (All-atom force fields) 中,体系的力点与分子中的全部原子一一对应,质量集中在原子核上。也就是说,力点与原子核的位置或原子的质心位置重合。简单来说,全原子力场中,分子由其组成原子为质点的集合构成。全原子力场的优点是具有更高精确度,可以更好地描述生物分子体系的结构与性质。但是,全原子力场的计算量大,模拟效率低。因此,在计算资源受到限制时,模拟者必须减少模拟体系的规模或缩短模拟体系的实际演化时间,影响模拟效果。
创建分子结构
AuToFF支持2D建模和3D分子显示,用户可以通过2D建模的方式建立自己所期望的分子构型。同时,也支持多种结构文件(.mol/.pdb/.xyz/.mol2)的导入识别。

图1.1 创建分子结构
AuToFF也内置了分子结构库(电解液分子、水分子、维生素分子、碳水化合物、酶、氨基酸分子等)。

图1.2 选取分子结构
用户在下拉框选择 选取分子结构 即可进入分子结构数据库选择常见分子。

图1.3 分子结构库
此外,也可通过SMILES字符串解析小分子化学结构并建模。点击 import图标 即可进入SMILES输入框,输入SMILES字符串,继而自动生成3D分子结构

图1.4 SMILES图标

图1.5 SMILES输入框
根据力场选择原子类型
目前支持的全原子力场包括GAFF、GAFF2、OPLS-AA、CGenFF、CVFF……,通用力场包括UFF、DREIDING。此外,针对特定的水模型支持多种力场函数类型表示,包括OPC、OPC3、SPC、SPC/E、SPC/Eb、TIP3P、TIPS3P、TIP3P-FB、TIP4P、TIP4P/Ew、TIP4P/2005、TIP4P/ICE、TIP4P/epsilon、SPC/E-HW、TIP3P-HW、TIP4P/2005-HW。用户也可选择多种charge类型,包括普适原子电荷1.14*CM1A、1.14*CM1A-LBCC、AM1BCC、MMFF94、XTB-RESP、CM5、1.2XCM5、QEq和AI电荷GNN-RESP等。
若用户利用量化优化后的结构进行力场拓扑文件生成,想用该结构的键长键角参数,即可选中左下角 使用当前结构的键长/键角 按钮,程序将自动保留原始输入结构的键长键角参数。
此外,AuToFF可实现mSeminario方法进行力场参数拟合,点击 力场参数拟合 按钮,用户仅需上传量化计算得到hessian文件即可进行参数拟合,hessian文件可支持BDF,gaussian,orca,XTB软件格式。

图1.6 力场参数拟合

图1.7 根据力场选择原子类型
Note
OPLS力场的势函数被严格限制于最常见的势函数形式:共价键伸缩势和键角弯曲势采用谐振子势函数,二面角扭曲势只包括Fourier展开式的前三项,van der Waals相互作用采用Lennard-Jones 12-6 势函数,静电相互作用采用库伦是函数。同时,所有力点位于原子(核)上,不考虑力点偏离原子中心。在计算分子内非键相互作用时,完全排除1-2和1-3相互作用,但只排除50%的1-4相互作用。计算交叉项的van der Waals相互作用时,采用Lorentz-Berthelot混合规则计算Lennard-Jones势函数。
在描写水分子的力场中,TIP3P是全原子力场,质点、力点、电荷点重合。相反,TIP4P、TIP5P等,力点的数目超过质点的数目或原子数。选择力点与分子中各个原子核的位置重叠,可以在模拟中省去重新分布受力和力矩的计算。
在模拟生物分子的水溶液时,OPLS力场应与TIP4P或TIP3P水分子模型搭配,可以取得更好的模拟效果。
CGenFF力场是CHARMM的通用力场,多了一项Urey-Bradley相互作用势,以弥补键角弯曲势的不足。
在DREIDING力场中,参与分子内或分子间相互作用的所有原子,都被按其成键的杂化类型或几何构型进行系统分类,同种类型的原子参与相互作用性质相同,不同种类型的原子参与相互作用性质不同。DREIDING力场在计算结合能和分子相互作用时的精度更高,但一些过渡金属元素相关参数存在缺失。
UFF力场比DREIDING力场有更广泛的适用范围,是对DREIDING力场的发展。
GNN-RESP电荷是基于机器学习的第一性原理精度的RESP原子电荷。
生成拓扑文件
用户建立完分子结构,选择相应的力场,进而生成拓扑文件,也可进行力场参数的修改。最后,选择计算软件从而生成相应的输入文件,支持GROMACS、LAMMPS、AMBER、MOLTEMPLATE、OPENMM、TINKER、CHARMM、GEAR、RASPA、NAMD、GOMC、GAUSSIAN、ORCA软件格式输出,其中GAUSSIAN、ORCA格式为QM/MM输入参数生成,若想生成BDF软件的QM/MM输入参数生成,用户选择AMBER软件下载拓扑文件,格式即可兼容。AuToFF可以有效帮助用户解决力场的选择、参数的生成、复杂体系的建模等多种分子模拟过程种遇到的困难,AuToFF可以有效降低这些使用门槛,可以极大的扩大分子动力学模拟的用户群体。

图1.8 生成拓扑文件
Note
AuToFF程序所提供的力场单位说明如下:
针对分子间相互作用力参数中的范德华半径单位是nm,势阱深度单位是kJ/mol
针对键伸缩能中参数1单位是nm,参数2单位是kJ/mol/nm^2
联合原子力场
联合原子力场 (united-atom force fields) 与碳原子直接键连的氢原子的相对原子质量被重叠到碳原子上,形成一个联合原子的整体,同时,其他原子对氢原子的相互作用也被叠加到联合原子上,从而可以大幅度降低力场的复杂性,减少势参数,减少计算非键相互作用的数量和计算时间。在联合原子力场中,力点数少于原子数,是分子的不完全表述。 目前支持的联合原子力场包括TraPPE-UA、PDMS、OPLS-UA、OPLS-UA/BCI、Dreiding-UA、TraPPE-UA+CL&P、TraPPE-UA+OPLS-2009IL、TraPPE-UA+OPLS-2009IL/0.8、OPLS-UA+CL&P、OPLS-UA+OPLS-2009IL、OPLS-UA+OPLS-2009IL/0.8、PDMS+TraPPE-UA
Note
TraPPE力场中所有键长被约束在平衡位置,不允许振动,不需要共价键伸缩势。因此,成键相互作用只包括键角弯曲势、二面角扭曲势和离面弯曲势三种类型。
OPLS-UA力场中,所有与碳原子成键的氢原子不直接出现在力场中,而是隐含在与碳原子的力场参数中。
同全原子模块操作方法一样,首先分子的2的建模/导入分子结构,产生3d结构;然后,选择联合原子力场,点击 下一步 程序根据力场自动选择划分原子类型,分配力场参数,生成该分子的力场拓扑文件。

图2.1 创建分子结构
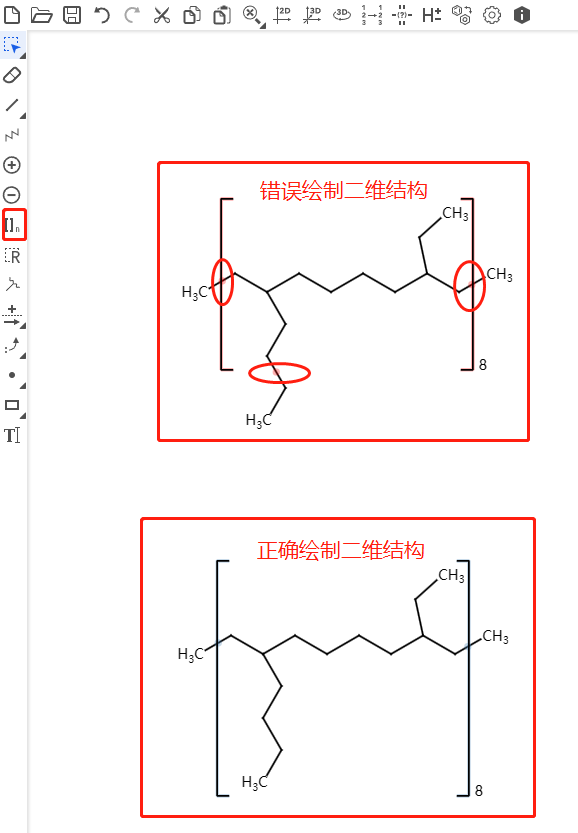
以下图壬烷为例,简化了原子类型,两端的CH3基团当作一个联合原子整体,中间的CH2基团作为一个整体。

图2.2 根据力场选择原子类型
点击 下一步 即可合理产生力场参数,最终生成对应软件格式的力场拓扑文件。

图2.3 生成拓扑文件
离子液体
目前支持的离子液体相关力场包括CL&P、OPLS-2009IL、Merz和Haiyang Zhang开发针对不同的水模型体系下的金属离子的LJ势函数。具体包括有CL&P、OPLS-2009IL、OPLS-2009IL/0.8、OPLS-IONS_2006、Madrid-2019、Merz/IOD/OPC、Merz/IOD/OPC3、Merz/IOD/SPCE、Merz/IOD/TIP3P、Merz/IOD/TIP3P-FB、Merz/IOD/TIP4P-Ew、Merz/IOD/TIP4P-FB、Merz/IOD/All_TIP3、Merz/HFE/OPC、Merz/HFE/OPC3、Merz/HFE/SPCE、Merz/HFE/TIP3P、Merz/HFE/TIP3P-FB、Merz/HFE/TIP4P-Ew、Merz/HFE/TIP4P-FB、Merz/CM/OPC、Merz/CM/OPC3、Merz/CM/SPCEMerz/CM/TIP3P、Merz/CM/TIP3P-FB、Merz/CM/TIP4P-EwMerz/CM/TIP4P-FB、HaiyangZhang/a99SB-disp、HaiyangZhang/OPC、HaiyangZhang/OPC3、HaiyangZhang/SPCE、HaiyangZhang/SPCEb、HaiyangZhang/TIP3P、HaiyangZhang/TIP3P-FB、HaiyangZhang/TIP4P-2005、HaiyangZhang/TIP4P-D、HaiyangZhang/TIP4P-Ew、HaiyangZhang/TIP4P-FB、SMALL/OH-、CRYSTAL/CaCO3、PolyOxoMetalates/VOx、PolyOxoMetalates/FeCl4、PolyOxoMetalates/Uranyl/GW。
首先是,创建分子结构,包含三种方式,同全原子力场操作。

图3.1 创建分子结构
第二,根据力场选择原子类型,自动匹配原子类型。

图3.2 根据力场选择原子类型
第三,生成对应的力场参数,产生力场拓扑文件。

图3.3 生成拓扑文件
碳材料
目前碳材料模块支持五种碳材料的建模,包括富勒烯、石墨烯、石墨炔、单壁碳纳米管、多壁碳纳米管。

图4.1 创建石墨烯结构
此外,搭建好碳材料基础结构后,选择 生成3d结构视图 后,再点击 官能团修饰 选项还可实现官能团修饰功能(官能团类型包括桥氧键、醚键、羟基、氨基、羧基、硝基、羰基)。

图4.2 石墨烯结构上随机加入桥氧键修饰
金属/共轭有机框架
目前本模块搭载了金属/共轭有机框架结构数据库,1)金属有机框架结构库:即MOFs材料,包含九种数据库,如CPO-27、IRMOF、M2、MIL、PCN、ZIF、CoRE、UiO等。2)共轭有机框架结构库:即COFs材料,包含CURATED-COFs和CoRE-COF_DT613-v6.0两类数据库。或者通过用户上传cif、vasp、gjf、car文件格式导入相应结构。支持全原子力场以及针对MOFs/COFs材料特定力场,如UFF4MOF、UFF4MOFII、ZIF-FF、ZIF-8等。

图5.1 金属/共轭有机框架界面
其中 根据力场选择原子类型 界面的电荷计算方法,其中包括Eqeq、EEM、GMP-Qeq、MEPO-Qeq、m-CBAC,还额外增加了高精度的机器学习预测MOF和COF材料的REPEAT和DDEC电荷,即packmof-MOF-REPEAT、packmof-COF-DDEC。

图5.1 金属/共轭有机框架——选择电荷类型
晶体材料
晶体材料是一类周期性结构,内部原子、离子或分子在三维空间内按照一定的规律排列,具有长程有序性。目前本模块搭载了多种晶体结构数据库,方便用户直接导入结构模型,包括有机分子晶体、锑化物合金、金属互化物、沸石、陶瓷、氧化物、硫化物、硫酸盐、氮化物、碳化物、碳酸盐、卤化物、氢氧化物、粘土等。也可通过用户上传cif、vasp、gjf、car文件格式导入相应结构。此外,针对晶体材料支持的力场包括ClayFF、CVFF/harmonic、CVFF/morse、CVFF_aug/harmonic、CVFF_aug/morse、Interface/PCFF、Interface/CHARMM、FCC-Metal、Metal、TraPPE-zeo、CRYSTAL/Silica、CRYSTAL/TiO2、CRYSTAL/CaCO3、PCFF、CFF91、COMPASS。

图6.1 晶体材料模块界面

图6.2 晶体材料结构库
Note
COMPASS力场是由上海交通大学孙淮教授课题组开发的第一个出自量子力学从头计算的新一代力场。比较CVFF、PCFF以及Universal等其他的力场,COMPASS力场的原子信息,能量和尺度参数等较全,可以应用于有机分子、无机分子和聚合物等多类复杂体系,甚至能准确得到凝聚态体系的物理图景和各种宏观性质,使模拟计算具有高精度的预测能力。使用该力场可以在很宽的温度和压力区间内准确得到体系中各粒子的振动情况及热物理性质,也可以用来研究具有表面或存在共混现象的比较复杂的体系,这是其他力场难以实现的。
PCFF力场更适用于聚合物一类的有机材料,以及包含二十种金属,也可运用在糖类、脂类和核酸体系中。
CVFF 力场又称一致共价力场,更适用于计算蛋白质,多肽等生化大分子有机体系。
聚合物建模
目前本模块支持均聚物、嵌段共聚物、无规共聚物建模、2D建模,此外还提供了较为完备的聚合物单体库,方便用户建立聚合物分子。本模块可选择的力场包括有:GAFF、GAFF2、GAFF2_mod、OPLS-AA/L、OPLS-AA/M、L-OPLS、OPLS-PFPE、OPLSR-PEO、CGenFF、Tripos、SYBYL、UFF、Dreiding、CVFF/harmonic、CVFF_aug/harmonic、CL&P、OPLS-2009IL、OPLS-2009IL/0.8、Interface/CHARMM、FCC-Metal、Metal、SMALL/EPM2、SMALL/IFF、SMALL/OH-、ClayFF。
均聚物
均聚物是由同一种单体通过聚合反应形成的聚合物。由于其结构的单一性,均聚物通常具有均匀的物理和化学性质,便于预测其性能和行为。均聚物的例子包括聚乙烯(PE)、聚丙烯(PP)和聚苯乙烯(PS)等。

Note
勾选 随机数种子 选项,程序会自动随机地生成聚合物结构。当然对于单一单体而言,确定了两个连接点,实际上就没有随机性了。但是有多种组分的时候,随机性就会发生作用了。
嵌段共聚物
嵌段共聚物是由两种或多种不同的单体段交替排列而成的聚合物。这种结构使得嵌段共聚物能够展现出独特的物理化学性质,例如自组装行为和相分离特性,广泛应用于材料科学和生物医药领域。

无规共聚物
无规共聚物是由两种或多种不同单体随机聚合而成的聚合物。这种随机排列的结构使得无规共聚物的性质介于均聚物和嵌段共聚物之间,具有一定的多样性和灵活性。现已支持两种方法构建无规聚合物链,分别是概率和竞聚率。当选择概率方法构建无规共聚物时,即给出每一个单体在下一步反应中连接到环上的可能性,并可以用于模拟浓度。对话框会出现n*n矩阵,该矩阵是按行来进行读入的。下图中,第一行表示,在聚合物链中,当上一个单体是对苯的时候,与之相连的下一个单体有50%的可能性是对苯,50%的可能性是噻二唑;第二行表示,当上一个单体是噻二唑的时候,与之相连的下一个单体有50%的可能性是对苯,50%的可能性是噻二唑。默认值是0.5,对于我们要求的组分比例为50:50来说刚好合适。

当选择竞聚率方法构建无规共聚物时,即用与实验合成相类似的方法测定共聚物成分,要求输入反应物浓度和多种连接选择的速率常数。竞聚率是反映共聚合反应特性的一个重要参数,表征单体的相对活性以及单体自聚和共聚倾向的大小。

Note
当想得到一个具有精确单体重复单元比例的聚合物时,用户需勾选 强制浓度
2D建模
2D建模功能允许用户在二维平面上构建和可视化聚合物的结构,便于进行初步设计和分析。为了寻找更便捷、更高效的聚合物建模方法,AuToFF上线了聚合物2D建模功能,用户仅需两步即可建立想要的聚合物结果,分别为:首先,跳转至marvin网站绘制聚合物结构;然后,复制mrv格式字符串到下方文本输入框生成3D结构

Note
marvin网站绘制2d结构确定重复单元时一定要框选所有的原子,将所有原子包含进去,不能出现红点标记,否则将会识别读取有误
生物大分子
本模块支持多糖分子的GLYCAM_06j力场,能够准确描述糖链的几何结构和相互作用,适用于复杂的糖类分子,包括寡糖和多糖。此外,本模块还支持蛋白质分子的FF19SB力场,在AMBER力场的基础上进行了优化,特别是针对氨基酸的侧链和主链的参数进行了改进,以提高对蛋白质结构和性质的描述能力。
多体势
多体势目前AuToFF涵盖多种类型,如:EAM、MEAM、SNAP、aSNAP、table、Tersoff。支持元素如下图:

反应力场
反应力场用来研究化学反应,允许化学键的断裂和生成,体系中各原子间也没有连接性,而是通过计算任意两个原子间的键级(Bond Order,BO)来确定当前时刻的连接性。目前反应力场模块支持COMB3、ReaxFF,AuToFF支持元素如下图:

粗粒化力场
目前支持全原子——Martini3粗粒化力场映射。程序根据全原子轨迹数据自动提供粗粒化映射方案,从而将全原子坐标转换为粗粒化坐标,匹配键合构象的映射分布。

图11.1 全原子——粗粒化力场映射关系
QM/MM建模
想要实现QM/MM输入参数生成可在全原子力场模块中依次实现创建分子结构——根据力场选择原子类型——生成拓扑文件,在最后 选择计算软件 选择量化Gaussian,ORCA和Amber软件即可,Amber对应BDF软件格式。
