分子动力学模拟(MD)
本章将介绍分子动力学相关知识。
分子动力学模拟(MD)
分子动力学模拟是一种通过模拟体系原子和分子运动轨迹研究体系微观和宏观性质的计算机模拟方法,是研究分子体系的重要手段。根据经典力学,只要给定分子体系中原子和分子之间的相互作用,就可以利用牛顿运动方程可以求解分子或原子的运动轨迹,然后使用一定的统计方法计算出系统的力学、热力学、动力学性质。在分子动力学中首先将由N个粒子构成的系统抽象成N个相互作用的质点,每个质点具有坐 标(通常在笛卡尔坐标系中)、质量、电荷及成键方式,按目标温度根据Boltzmalm分布随机指定各质点的初始速度,然后根据所选用的力场中的相应的成键和非键能量表达形式对质点间的相互作用能以及每个质点所受的力进行计算。接着依据牛顿力学计算出各质点的加速度及速度,从而得到经一指定积分步长(通常1fs)后各质点新的坐标和速度,这样质点就移动了。经一定的积分步数后,质点就有了运动轨迹。设定时间间隔对轨迹进行保存。最后可以对轨迹进行各种结构、能量、热力学、动力学、力学等的分析,从而得到感兴趣的计算结果。分子动力学模拟方法已被广泛应用于化学、物理、材料科学、生物学等学科领域。
MD模拟的统计力学基础
统计力学利用统计学方法和微观粒子的运动规律,研究多粒子体系的宏观性质,是现代物理学的支柱之一。利用统计力学解决热力学问题时,统计力学被称为统计热力学。宏观体系处于宏观静止状态,但其组成原子、分子处在不断的运动状态。在热力学,常用一组宏观可观测量描述宏观体系的状态。Gibbs提出,可以利用宏观可观察量作约束条件,定义具有不同统计特性的热力学体系,即热力学系综。重要的热力学系综包括:微正则系综(microcanonical ensemble, NVE),正则系综(canonical ensemble, NVT),巨正则系综(grand canonical ensemble, µVT),等温等压系综(isothermal–isobaric ensemble, NPT),等焓等压系综(isoenthalpic-isobaric ensemble, NPH)。
在NVE系综中,体系的粒子数(N)、体积(V)、总能量(E)均保持守恒,对应与环境没有能量交换、粒子交换的孤立体系。在NVT系综中,体系的粒子数(N)、体积(V)、温度(T)保持守恒,在MD模拟中通过体系与恒温热浴的能量交换实现温度守恒。常用控温算法有Nosé–Hoover控温、Berendsen控温、Andersen控温、Langevin控温等。在NPT系综中,体系的粒子数(N)、压强(p)、温度(T)均保持守恒,但体系体积可发生变化。实验室化学反应常在恒压下进行,NPT系综常被用于与化学反应相关的研究。µVT系综对应开放体系,不但与热浴存在能量交换,还与环境进行物质交换,体系的能量和粒子数不守恒,但系综与环境保持统计平衡。在NPH系综中,保持系统的粒子数N、压力P和焓值H都不变。然而,模拟时要保持压力与焓值为固定值,有一定难度。事实上,这种系综在实际的分子动力学模拟中很少见。
根据统计热力学,可以利用经典力学或量子力学计算系综平均,得到体系的热力学性质等。
分子内和分子间相互作用
分子间力是分子间相互作用产生的力,包括分子与近邻粒子(原子或离子)之间相互作用产生的引力或斥力,影响体系的物理化学性质。分子内力是同一分子中的原子通过共用电子对产生相互作用力,影响体系的化学性质。分子间力通常小于分子内力,共用电子引起的共价键远强于分子间相互作用力。分子体系相互作用由分子间力和分子内力组成。
在MD模拟中,常用分子间相互作用势U(r)来表示分子间相互作用,r为分子间距离。因此,分子间相互作用势决定了物质的结构性质和物理化学性质。Van der Waals在研究实际气体和液体性质时,考虑了分子间相互作用力,提出在分子间距离r较小时分子间存在斥力,随着距离r的增大分子间的斥力逐渐被引力所取代,推导得到实际气体van der Waals状态方程。
分子间相互作用力分为分子间排斥力和吸引力两个部分。分子间斥力的本质是遵循Pauli不相容原理的波函数发生重叠时的排斥力。分子间引力由分子电子分布中心与原子核正电中心分离、色散等效应引起。在Lennard-Jones势函数中,近似于一对非键合原子或分子之间的非静电相互作用的势能,分子间引力随 \(r^{-6}\) 变化,分子间斥力随 \(r^{-12}\) 变化(图1),数学函数可表示为,
其中,r=σ对应体系势能为0的位置,ε是势阱的深度。对于复杂体系,难于利用量子力学理论精确计算U(r),也难于用实验测量分子间相互作用力。
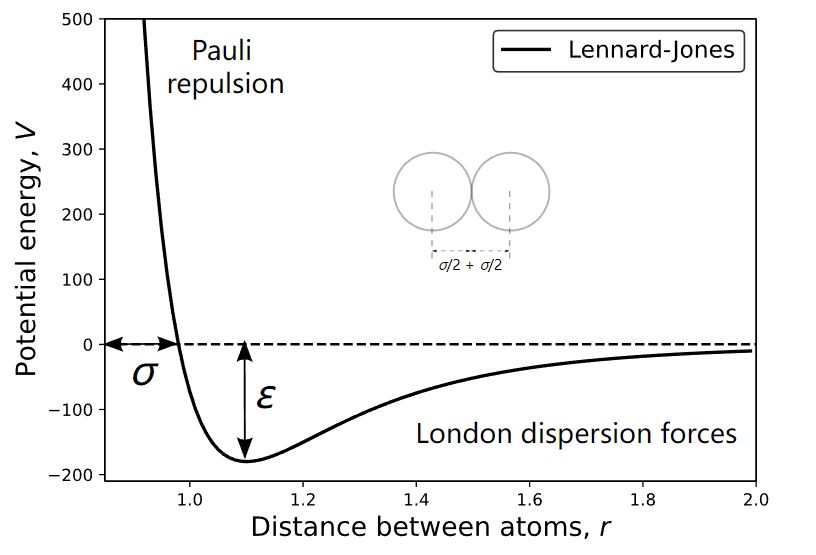
图 1 分子间相互作用Lennard-Jones势函数U(r)与分子间距离r的关系
分子间相互作用模型
力场模型是描述体系势能与粒子坐标关系的一组经验势函数,包括势能函数形式和力场参数两个部分。常用的经典力场模型有DREIDING、UFF、OPLS、CVFF、AMBER、GROMOS、CHARMM、COMPASS等。力场参数可以通过从头计算或半经验量子化学计算得到,也可以通过物理化学实验数据(如中子衍射、X射线衍射、电子衍射、核磁共振谱图、红外光谱、拉曼光谱等)拟合得到。分子体系总能量包括成键相互作用和非键相互作用两个部分,
其中,\(E_{bonded}\) 是分子内相互作用,包括键长伸缩、键角弯曲、二面角扭曲、交叉项等作用,
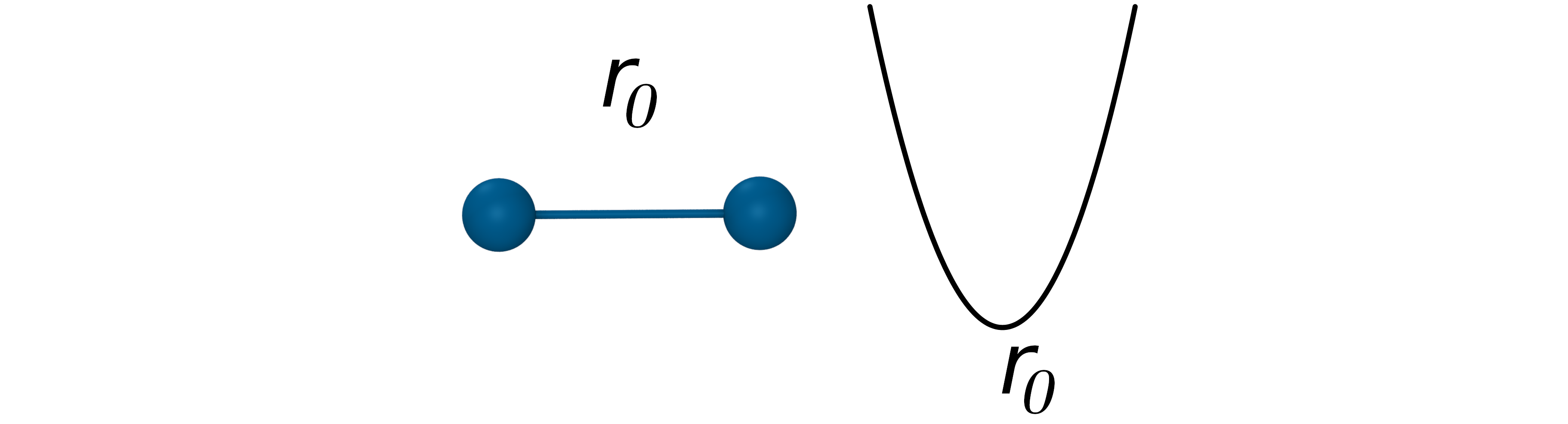
键伸缩能:表示分子种原子间共价键的相互作用,近似为谐振子势函数
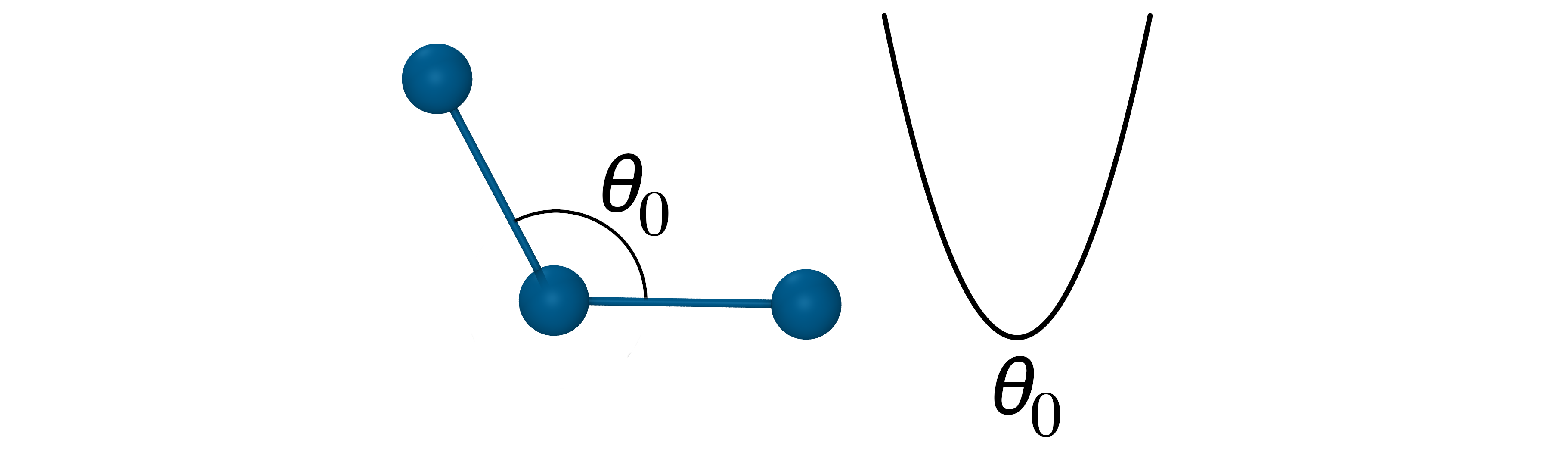
键角弯曲能:表示三个原子所形成的键角变化所形成的能量,常见的完整函数形式一般使用的是谐振子势函数
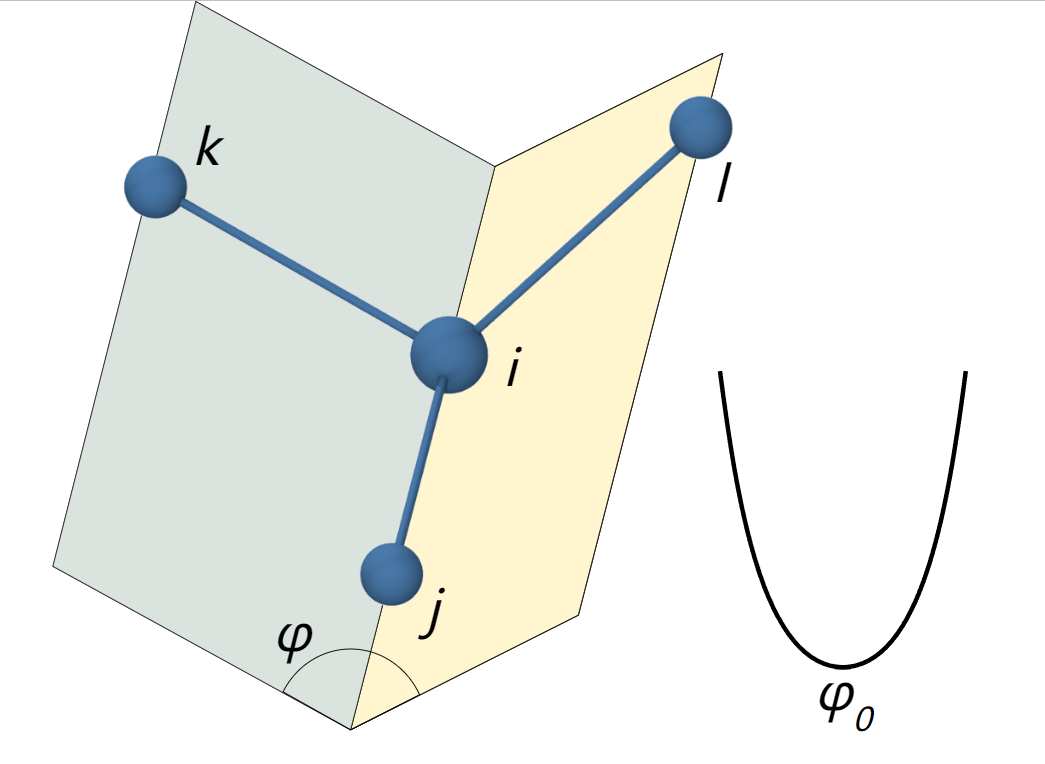
二面角扭转能:表示四个键合原子,扭转角是围绕中间两个原子之间共价键的旋转角,与键伸缩能和键角弯曲能不同的是,二面角扭转能一类具有多个最低点和最高点,因此采用周期性势函数进行描述

赝扭曲势(improper):表示四个键合原子组成的反常扭转势,其中中心原子i与3个外围原子j、k和i相连。主要用来保持分子结构的平面性
\(E_{nonbonded}\) 是非键相互作用,包括长程静电力和van der Waals力两部分,
在MD模拟中,常用Lennard-Jones势近似 \(E_{vdW}\) 。
MD模拟的积分算法
实际体系中,描述粒子间相互作用的势能函数复杂,难以求解牛顿运动方程的解析解。因此,在MD模拟过程中采用数值积分的方法求解体系的运动方程,有关常用数值积分算法如下,
Verlet算法:利用Taylor展开粒子的位置坐标,
上述两式相加得到Verlet算法的基本公式,
利用Verlet算法计算 \(t+\delta t\) 时刻的粒子位置,需要t时刻的粒子位置和加速度、以及 \(t-\delta t\) 时刻的粒子位置。该算法计算简单,不直接计算粒子的速度,但算法精度不高。
Leap-frog算法:首先计算 \(t+\frac{1}{2} \delta t\) 时刻的粒子速度,
然后,计算 \(t+\delta t\) 时刻的粒子位置,
Leap-frog算法方法虽然直接计算体系的粒子速度,但体系的粒子速度和位置不同步。 \(t\) 时刻的粒子速度近似为,
Velocity Verlet算法:具有更高的计算精度,体系的粒子位置、速度分别表示为,
Beeman’s算法:基于Verlet算法改进体系的粒子位置和速度分别为,
Beeman’s算法的计算精确度得到了极大的提高,但计算成本也相应提高
分子力场类型
分子力场根据量子力学的波恩-奥本海默近似,一个分子的能量可以近似看作构成分子的各个原子的空间坐标的函数,简单地讲就是分子的能量随分子构型的变化而变化,而描述这种分子能量和分子结构之间关系的就是分子力场函数。一般而言,分子力场函数由以下几个部分构成:
键伸缩能:构成分子的各个化学键在键轴方向上的伸缩运动所引起的能量变化
键角弯曲能:键角变化引起的分子能量变化
二面角扭曲能:单键旋转引起分子骨架扭曲所产生的能量变化
非键相互作用:包括范德华力、静电相互作用等与能量有关的非键相互作用
交叉能量项:上述作用之间耦合引起的能量变化
不同的分子力场会选取不同的函数形式来描述上述能量与体系构型之间的关系。
GAFF
GAFF(Generation Amber Force Field) [1] 力场是普适型有机小分子力场,函数形式和AMBER力场相同,与AMBER力场完全兼容。
AMBER
AMBER [2] 力场是传统力场之一,主要适用于蛋白质和核酸体系、多糖。
CVFF
CVFF(Consistent Valence Force Field) [3] 属传统力场。适应于有机小分子和蛋白质体系。扩展后可用于某些无机体系的模拟,如硅酸盐、铝硅酸盐、磷硅化合物等,主要用于预测分子的结构和结合自由能。
Bond:
Morse:
Harmonic:
OPLS
OPLS(Optimized Potentials for Liquid Simulations) [4, 5, 6] 力场主要是适用于多肽、蛋白、核酸、有机溶剂等液体体系。
MMFF
MMFF(Merck Molecular Force Field)是小分子力场。
UFF
UFF(Universall Force Field) [7, 8] 力场覆盖了周期表中所有元素,应用最为广泛。
Bond:
Harmonic
Morse
Angle:
Torsion:
LJ:
Dreiding
Dreiding [9] 是普适型力场,但支持的元素有限,并非涵盖整个周期表。可以用于有机、生物、主族无机分子。结构、结合能的计算结果精度一般。
Bond:
Harmonic
Morse
Angle:
Torsion:
LJ:
LJ rules:
However, Dreiding-X6:
PCFF
PCFF [] 基于CFF91,适用范围做了扩展,主要用于聚合物和有机材料,也能用于无机材料,还有糖、核酸、脂的参数。
CFF
CFF(Consistent Family of Forcefield) [10, 11] :包括CFF91和CFF95。适用面很广,涵盖有机无机小分子、聚合物、多糖和生物大分子,还支持金属。
CFF91
CFF91 [12] 主要用于模拟有机小分子、蛋白质以及小分子-蛋白质之间的相互作用。
CFF93
CFF93 [10] 在CFF91的基础上进行了改进,增加了对更复杂分子系统的支持。该力场提供了一致的参数集,能够有效地描述分子间的相互作用,适用于计算化学、材料科学和生物物理学等领域的研究。
ClayFF
ClayFF [13] 力场是一种通用力场,适用于水合和多组分粘土体系及其与水溶液界面的模拟。
GROMOS-53A5 and 53A6
GROMOS(Groningen Molecular Simulation) [14] 力场是联合原子力场,用于生物分子模拟的最重要分子力场之一。
Covalent Bond Interactions
Covalent Bond-Angle Interactions
Improper Dihedral-Angle Interactions
Torsional Dihedral-Angle Interactions
Van der Waals Interactions
Electrostatic Interactions
CHARMM
CHARMm力场(Chemistry at Harvard Macromolecular Mechanics)也是传统力场之一。适用于各种分子性质的计算和模拟。对于从孤立的小分子到溶剂化的生物大分子体系的多种模拟体系都可以给出较好的结果,但不适合于有机金属配合物。
CGenFF
CGenFF(CHARMM General Force Field) [15] 用于药物类小分子,也可视为通用有机小分子力场。可结合其它CHARMM全原子力场使用。
Intramolecular(internal, bonded terms)
Intermolecular(external, nonbonded terms)
分子动力学模拟软件
常用的分子动力学模拟模拟程序有LAMMPS、AMBER、CHARMM、GROMACS和OpenMM等,接下来将介绍AuToFF工具所支持生成如下分子动力学模拟软件的输入文件,未来将支持更多的分子动力学模拟软件。
LAMMPS
LAMMPS程序(Large-sale Atomic/Molecular Massively Parallel Simulator)是由美国Sandia国家实验室开发的开源经典力学MD模拟程序,侧重于材料领域的模拟研究。LAMMPS程序在模拟固态材料(金属、半导体)、柔性物质(生物分子、聚合物)、 粗粒度介观体系等方向具有广泛的应用。持续内置多种原子间势(力场模型),可以实现原子、聚合物、生物分子、固态材料(金属、陶瓷、氧化物)、粗粒度体系的建模和模拟。该程序既可以模拟二维体系,也可以模拟三维体系,可以模拟多达数百万甚至数 十亿粒子的分子体系,具有模拟效率高、计算时间短等优点。LAMMPS程序具有良好的用户界面,用户可以自由修改或扩展新的力场模型、原子类型、边界条件等以满足课题研究需求。LAMMPS可以在单个处理器的台式机和笔记本本上运行且有较高的计算效率, 但是它是专门为并行计算机设计的。他可以在任何一个安装了C++编译器和MPI的平台上运算,这其中当然包括分布式和共享式并行机和Beowulf型的集群机。LAMMPS的部分功能还支持OpenMP多线程、矢量化和GPU加速。通常意义上来讲,LAMMPS是根据不同的边界条件和初始条件对通过短程和长程力相互作用的分子,原子和宏观粒子集合对它们的牛顿运动方程进行积分。
GROMACS
GROMACS程序(GROningen MAchine for Chemistry Simulation)是一款集成了高性能分子动力学模拟和结果分析功能的免费开源软件,高度优化的代码使GROMACS成为迄今为止分子模拟速度最快的程序。GROMACS程序可以模拟具有数百至数 百万个粒子的系统的牛顿运动方程。GROMACS旨在模拟具有许多复杂键合相互作用的生化分子,例如蛋白质,脂质和核酸。另外,GROMACS能够⾮常快速地计算⾮键作⽤,因此也可⽤于⾮⽣物体系,如聚合物、⼀些有机物、⽆机物等。 GROMACS于上世纪90年代初诞生于哥廷根大学Berendsen实验室,其开发初衷是发展一个并行的分子动力学软件,初版功能主要基于van Gunsteren和Berendsen开发的串行动力学软件GROMOS。虽然与GROMOS有很深的渊源,GROMACS诞生之后两个软件各自独立发展,分别由Berendsen实验室和van Gunsteren维护和开发,在功能和特性上也渐趋不同。van Gunsteren实验室侧重于与GROMOS同名的力场的开发,Berendsen实验室则在动力学软件本身的开发尤其是性能提升方面取得了很多进展。从2001年开始,GROMACS的开发维护工作由瑞典KTH皇家技术学院的Science for Life Laboratory主导。GROMACS主体代码使用C语言,近年来正逐步过渡到C++,代码开源。在发展历程中GROMACS一直强调性能优化,其运行效率尤其是单机计算效率在多个benchmark中明显优于几个主流同类软件。时至今日,高度优化的计算性能和代码的开放性为GROMACS赢得了众多的用户,使之成为目前生物系统分子动力学模拟领域中最常用的软件。
AMBER
Amber程序是一款旨于模拟蛋白质、核酸、糖等生物大分子的分子动力学软件,包括拥有一套模拟生物分子的分子力场和分子动力学模拟程序包。Amber分子动力学模拟软件由AmberTools22和Amber22组成,AmberTools22(开源的)是由几个独立开发的软件包组成,这些软件包可以单独使用,也可以与Amber22一起使用。该套件还可以用于使用显式水模型或广义Born溶剂模型进行完整的分子动力学模拟,包含:1. Leap:用于准备分子系统坐标和参数文件,有两个程序:xleap:X-windows版本的leap,带GUI图形界面。tleap:文本界面的Leap。2. Antechamber:加载生成部分缺失的力学参数文件。3. Sander:分子动力学模拟程序,被称做AMBER的大脑程序。4. Ptraj:分子动力学模拟轨迹分析程序。Amber22(商业版)软件包通过添加PMEMD程序在AmberTools22上构建,支持GPU加速,该程序类似于AmberTools中的sander(分子动力学)代码,但在多个CPU上提供了更好的性能,并在GPU上显着提高了速度。
Moltemplate
Moltemplate是LAMMPS官方支持的建模工具之一,既可以建立粗粒化模型,也可以建立全原子模型。Moltemplate创建了一种简单的文件格式来存储分子定义和力场,即模板LT,其中LT文件包含与特定分子有关的所有信息(包括坐标、拓扑、角度、力场参数、shake约束、k空间设置、甚至组定义)。 Moltemplate支持使用ATB分子数据库(https://atb.uq.edu.au),下载适应于目标分子的LT文件或手动创建LT文件;也支持各种现有力场类型,即:OPLS , AMBER (GAFF,GAFF2), DREIDING , COMPASS , LOPLS (2015), EFF , TraPPE (1998) , MOLC , mW , ELBA (water), oxDNA2。 Moltemplate可以复制分子,对其进行自定义,然后用它作为构建更大、更复杂分子的基础。构建后,可以自定义单个分子和亚基(原子和键,以及可以插入、移动、删除或替换子单元)。Moltemplate支持所有LAMMPS力场样式以及几乎所有原子样式。Moltemplate目前可与以下软件进行联用:VMD、PACKOL、OVITO、CellPACK、VIPSTER、EMC和OpenBabel。
OpenMM
OpenMM是一个高性能的分子模拟工具包,既可以用作运行模拟的独立应用程序,也可以用作库被其他程序调用。OpenMM集合了极高的灵活性(自定义功能)、开放性和高性能(通过GPU加速以及AMD、NVIDIA和Intel集成GPU的优化实现了卓越的性能)、安装简单等众多优势, 使其在仿真软件中真正独树一帜。OpenMM软件架构设计更加模块化,也易于扩展。同时在底层的核心程序都使用C++编写,用户交互及力场参数的处理则都是采用了Python编写。既保证了计算性能的同时也使得使用上更加便捷易懂。目前,OpenMM大多用于生物体系的模拟当中,同时也支持CHARMM, Amber, Drude 等大多数常用力场。
Tinker
Tinker软件包是由Jay William Ponder教授开发的一个较完整的分子模拟软件包,涵盖分子建模、分子力学、分子动力学的计算,具有一些特殊的生物高分子特性。Tinker可以使用Amber (ff94, ff96, ff98, ff99, ff99SB), CHARMM (19,22,22 /CMAP), Allinger MM (MM2-1991和MM3-2000), OPLS (OPLS-UA, OPLS-AA), Merck分子力场(MMFF), Liam Dang的可极化模型,AMOEBA(2004, 2009, 2013, 2017, 2018)可极化原子多极矩力场,加入电荷穿透效应的AMOEBA+和新HIPPO (类氢原子间极化势)力场等几种常用参数集,未来还将集成更多的力场类型。
CHARMM
CHARMM程序(Chemistry at HARvard Macromolecular Mechanics)是一个广泛应用于多粒子系统的分子动力学模拟程序,具有一套全面的能量函数集,多种增强采样方法,并支持多尺度模拟,包括QM/MM, MM/CG和多种隐式溶剂模型。 CHARMM于70年代末诞生于Martin Karplus小组,其前身正是历史上首次尝试基于蛋白质结构计算能量所使用的程序。该程序最初基于Lifson的consistent force field (CCF),其后由Bruce Gelin和Andy MacCammon等发展,成为从结构到相互作用再到动力学模拟的一套方法。1983年正式发表文章。正是由于与蛋白质的计算机模拟发展史息息相关,CHARMM同时也是领域内历史最悠久、使用最广泛的一种力场的名称。数十年来,CHARMM软件及力场与生物大分子的动力学模拟方法一直同步发展,参与的开发人员来自世界各地,而主要贡献者多半曾是Martin Karplus的学生或博士后合作者。软件主要使用Fortran开发,现有代码量约百万行。由于参与软件开发的人员大部分同时也是算法本身的开发者,该软件集成了生物大分子动力学模拟领域的各种前沿算法。